










Background. Cyclooxygenase-2 (COX-2) enzyme over expression is reported in many human HCC cell line studies and is linked to tumor cell resistance to chemotherapy-induced apoptosis. We hypothesized that adding a COX-2 inhibitor would improve the therapeutic benefits in patients with HCC. COX-2 is often increased and involved in drug resistance and poor prognosis.
Method. Between January 2001 and December 2007, 15 patients with MDR-positive-HCC from 34 HCC patients based on tissue and serum liver of glypican-3 and fitting the preset eligibility criteria, were treated with a combination regimen with intravenous infusion of (5-FU) 750 mg once per week, 100mg/day cyclophosphamide (Endoxan) and 400 mg/day celecoxib taken orally in divided doses, while the rest of the patients received only 5-FU and Endoxan. Twenty-one patients (62%) had liver disease associated with hepatitis C virus (HCV) and 5 patients with hepatitis B virus (62%). Results. We found that celecoxib reduced P-glycoprotein with activation of caspase-3 and marked regression of tumor sizes. Sera angiogenic factors (VEGF & bFGF) levels measurement in HCC patients indicated that, the sera levels of both angiogenic factors were reduced significantly (p < 0.05) after treatment. Based on the tumor markers AFP & Glypican-3, 11 of the patients had a PR (11/15), including 3 patients who had normalization of AFP, and four patients had CR (4/15).
Conclusions. These data suggest that the combination of 5-FU, Endoxan and Celecoxib is highly effective palliative regimen for patients with HCC with good performance status (score ≤ 3). The study suggests a framework for Celecoxib-based combination treatment of HCC.
The study of pathogenesis of hepatocellular carcinomas has shown that the molecular basis of the malignant phenotype is highly heterogeneous, and it is thought that several of the proteins that play a role in the progression of this disease remain to be identified.1,2 As a result of a search for genes that could play a role in hepatocellular carcinoma, Zhu, et al., reported that glypican-3 (GPC3) mRNA levels are significantly elevated in most hepatocellular carcinomas compared with normal liver and nonmalig-nant liver lesions.3,4 Also high expression of several genes involved in drug metabolism and inactivation, with P-glycoprotein and/or multidrug resistance gene 1 (MDR1) expression have been shown to be inversely related to the response to systemic chemo-therapy.5
Multidrug resistance, MDR, remains a major and difficult problem to resolve in the therapy of HCC. There is compelling evidence linking cyclooxygena-se-2 (COX-2) and carcinogenesis, Overexpression of COX-2 is associated with carcinogenesis in colorec-tal, prostate, and breast cancers, and in hepatocellu-lar carcinoma.6,7 The frequency of aberrant COX-2 expression increased gradually from chronic hepatitis, cirrhosis, to dysplasia.8 These findings indicate that COX-2 expression plays an important role in hepatic inflammation and malignant transformation of hepatocytes. A new class of COX-2 selective inhibitors (coxibs) preferentially inhibits the COX-2 enzyme thereby reducing side effects such as gastrointestinal ulceration and bleeding and platelet dysfunction caused by inhibiting COX-1. They inhibit the proliferation and induce apoptosis in cultured hepatocellular carcinoma cells, although these inhibitors are known to mediate effects through both COX-dependent and COX-independent mechanisms.9 Celecoxib induces cytochrome c release, activation of caspase-9 and caspase-3 and eventually apoptosis in hepatocellular carcinoma cells.10,11 Because COX-2 and P-glycoprotein proteins are expressed in hepato-cellular carcinoma12,13 they could affect the apoptotic pathways and cooperate in the development of the MDR phenotype, at least in human hepatocellular carcinoma. In recent years, inhibitors of COX-2, cele-coxib included, have been proposed in the treatment of hepatocellular carcinoma, and in fact, the inhibition of COX-2 was proved to possess antiproliferati-ve action.14 Preclinical studies have shown that celecoxib could overcome MDR1-mediated multidrug resistance and has significant antitumor activity against HCC cell lines.14 Here we report the efficacy and safety of a new strategy to improve survival of HCC patients by the addition of oral Celecoxib to definitive chemotherapy.
Materials and MethodsPatients and controlsBetween January 2001 and December 2007, thirty-four of 81 HCC patients who received 5-FU and cyclophosphamide (Endoxan) chemotherapy during the period were deemed to fit our eligibility criteria and were included in this prospective clinical trial in the Department of Internal Medicine, Faculty of Medicine, at Alexandria University. These criteria included cirrhosis or chronic hepatitis B [HBsAg (+), 5 (15%)] or hepatitis C infection [Anti-HCV (+), 21 (62%) and none 8 (23%)], hypervascular liver masses more than 2 cm, and either serum al-pha-fetoprotein of at least 500ng/mL. All 34 cancer patients were male, ages ranged from 40-75 years old, with histological confirmation of HCC or diagnosed by clinical criteria. They were Child-Pugh Class A or compensated Class B liver dysfunction [no refractory ascites or encephalopathy] 26/8, and renal function (serum Creatinine ≤ 2.0 mg/dL and urinary protein > 500 mg/24 h).
Fifteen of these 34 patients with MDR-positive-HCC were treated with a combination regimen with intravenous infusion of (5-FU) 750 mg once per week, 100mg/day cyclophosphamide (Endoxan) and 400 mg/day celecoxib taken orally in divided doses, while the rest (19 patients) received only 5-FU and cyclophosphamide (Endoxan). Patients had measurable disease (at least one non-bone tumor >2 cm) and were not candidates for surgical resection. This clinical trial was approved after Research and Ethical committee approval from Alexandria University was granted, with written consent from all patients. All patients were non-alcoholic. Standard pretreat-ment evaluation always included a complete medical history with physical examination and Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 2.15 Other eligibility requirements included no more than one prior antineoplastic chemotherapy and adequate organ function including hematologic (absolute neutrophil count [ANC] >1,200/mm3, platelet count ≥ 75,000/mL, prothrom-bin time ≤ 1.5 international normalized ratio [INR]), hepatic (serum bilirubin ≤ 3.0 mg/dL, and aspartate aminotransferase (AST) ≤ 7 times upper limit of normal, albumin ≥ 2.5 g/dL.
Treatment PlanThey were classified according to ultrasonography, computed tomography scanning, and clinical investigations16,17 into two major groups; group 1, comprised 15 patients and were treated with a combination regimen with intravenous infusion of (5-FU) 750 mg over 60 minutes on day 1 once per week, 100 mg/day cyclophosphamide (Endoxan) and celecoxib was taken continuously at a fixed dose of 200 mg orally twice a day without a break. Group 2, comprised 19 patients and were treated with combination of 5-FU ((750 mg in (5%) glucose solution) weekly, and Endoxan. The treatment was repeated every 21 days followed by a 7-day break. The leukocyte counts had to be > 1,000 cells/μL before starting the next cycle of treatment. All analyses were performed at the time of diagnosis and continued during the treatment follow up period. Treatment was continued for a maximum of ten cycles until one of the following criteria was met: disease progression, unacceptable toxicity or patient refusal.
Efficacy and safety assessmentPatients were seen on day 1 of every treatment cycle for a brief history, physical examination, assessment of adverse events, complete blood count, and renal and liver function tests. Assessment of tumor response was determined via the Response Evaluation Criteria in Solid Tumors (RECIST) cri-teria,18 after completion of the first two treatment cycles and every two to three treatment cycles thereafter by a computed tomography (CT) or magnetic resonance imaging (MRI) scan, or sooner if there were clinical signs of disease progression. The designation of stability or response required radiographic confirmation no fewer than 4 weeks following the initial determination.
Liver BiopsyLiver biopsies were performed using a Tru-Cut needle under ultrasound guidance. The tissue samples were collected in liquid nitrogen and snap-frozen at-80°C until cytosolic fraction extraction and RNA isolation were performed from tissue samples of all patients. Liver biopsies were obtained from both groups of patients at different time interval during the study course.
Response CriteriaTumor response evaluations were performed by physical examination, tumor markers (every cycle) and/or ultrasonography (every 9 weeks or every three cycles). A complete response (CR) was defined as the complete disappearance of all radiographic evidence of tumors and regression of all tumor sizes for a minimum of 6 weeks. A partial response (PR) was defined by a ≥ 20% reduction of the sum of the longest diameters of representative tumors lasting at least 6 weeks, or by a reduction of VEGF & bFGF by more than 50% lasting for more than 6 weeks. Progression was defined as an increase > 25% in tumor size, the appearance of any new lesion for more than 6 weeks.
Measurements of sera VEGF and bFGF levelsConcentrations of VEGF in the sera of all HCC patients and control subjects were determined with a commercially available Sandwich ELISA kit (Quantikine Human VEGF Immunoassay, R&D Systems, Minneapolis, MN) following the manufacturer’s guidelines. In each analysis, human recombinant VEGF165 was used as a standard and serially diluted according to the manufacturer’s information. VEGF levels were calculated from the best-fit curve through the points on the standard curve which determined by regression analysis. Sera bFGF levels were determined with a commercially available Sandwich ELISA kit (Accucyte kit, R) following the manufacturer’s guidelines. Samples were analyzed in duplicate. The amount of bFGF detected in each sample is compared to a bFGF standard curve which demonstrates an inverse relationship between optical density (O.D) and bFGF concentrations.
Measurement of serum GPC3 levelsSerum GPC3 levels were measured by using a sandwich enzyme linked immunosorbent assay (ELISA) method as described previously.4
Measurement of cytosolic PGE2 and VEGF levels in tumor biopsy liver tissuesCytosol was obtained by sonication of tumor liver tissues from all patients. In brief, frozen tissues were sonicated in four volumes of RIPA buffer (25 mmol/L Tris, pH7.4, 0.15 mol/L KCl, 1% NP-40, 5 mmol/L EDTA, 0.5% sodium deoxycholate, 0.1% SDS). Sonication was performed for five cycles of 30-s bursts at 1-min intervals on ice. The supernatant was collected after 5 min of centrifugation at 15 000 g at 4° C and assayed for the PGE2 and VEGF levels immediately. The total protein concentration in the supernatants was measured using Bio-Rad total protein assay (Bradford, Hercules, CA, USA). The PGE2 level in tissue cytosol was determined by using a commercial immunoassay kit (Prostaglan-din E2 ELISA Kit-Monoclonal, EIA) kit (Cayman Chemical, MI, USA). The procedure of the EIA provided by the manual was followed. The VEGF content of tissue cytosolic samples was quantified by an enzyme-linked immunosorbent assay (Quantikine Human VEGF Immunoassay, R&D Systems, Minneapolis, MN, USA). The assay employs the quantitative sandwich enzyme immunoassay technique using recombinant human VEGF and antibodies raised against the recombinant protein. Details of the assay have been described elsewhere.19,20 The assay exhibits no significant cross-reactivity with other angiogenic factors. All samples were assayed in duplicate.
Western blot analysisTotal proteins were extracted and evaluated by Western blot analysis, as previously described21 using each of the following primary antibodies: anti-COX-2 polyclonal antibody (Cayman Chemical); anti-P-glycoprotein (C219) monoclonal antibody (Calbiochem); anti-caspase-3 (E8) monoclonal antibody, and anti-GPC3 (Abcam, ab66596). Cytosolic extracts (25 μg protein) were subjected to 12.5% SDS-PAGE and transferred to a nitrocellulose membrane (Trans-Blot 0.2 μm; Bio-Rad). Western immuno-blotting analysis with mouse monoclonal anti-VEGF antibody (Monoclonal antibody mouse-anti-human VEGF was a product of Neomarker, Fremont, CA, USA), and rabbit monoclonal anti-b-actin (Sigma; 1:1,000 dilution) antibody was carried out using enhanced chemiluminescence assay (ECL kit, Amer-sham).
RNA isolation and reverse transcription-PCR analysisTotal RNA was prepared using Trizol (Invitrogen) according to manufacturer’s instructions. Reverse transcription was carried out with Superscript First-Strand Synthesis System for reverse transcription-PCR (RT-PCR; Invitrogen). As a control, duplicate cDNA synthesis reactions were done for each experiment without the addition of reverse transcriptase. 2 μg total RNA was reverse transcribed with MuLV reverse transcriptase. Sequences of the primers used for RT-PCR analysis of COX-2 and GAPDH are (Sense 5’-TGAAACCCACTCCAAACACACAG-3’; Anti-sense 5’- TCATCAGGCACAGGAGGAAG-3’) and (Sense 5’-ATGGCACCGTCAAGGCTGAG-3’; Anti-sense 5’-GCAGTGATGGCATGGACTGT-3’) respectively. Thirty-two cycles of amplifications were performed under the following conditions: at 95 for 2 min, at 94 for 45 s, at 56 for 45 s, at 72 for 45 s. The final extension step was performed by one cycle at 72 for 10 min. Reaction products were run by electrophoresis on 1.5% agarose gel for 30-40 min at 100 V in 0.5xTBE buffer, and visualized with ethidium bromide staining under UV light. Relative expression level of COX-2 was defined as optical density ratio against GAPDH as an internal control.
Statistical analysisThe primary end point was overall rate of disease control (response and stability). Our primary objective was to assess patient response rate, and our secondary objectives were to evaluate overall survival (OS), progression-free survival (PFS) and toxicity. Descriptive statistics were reported as proportions and medians. The PFS was measured from the first day of treatment until disease progression or date of death from any cause, and OS was measured from the first day of treatment until death from any cause. The data are presented as mean ± standard deviation for the number of experiments. Statistical significance (p < 0.05) between the experimental groups was determined by means of analysis of Fisher method of multiple comparisons. Statistical significance was taken as P < 0.05.
ResultsFifteen of the selected 34 patients with MDR-posi-tive-HCC (group 1) and median age of 56 years (range, 31 to 62 years) were treated with this regimen and had elevated serum GPC3 levels with values ranging 31.5-8120.6 ng/mL, and AFP levels were ≥ 900 and 1600 ng/mL. All patients were chemotherapy naive when starting the treatment. All 15 patients had no metastatic disease on presentation. Based on the tumor marker AFP, 11 of the patients had a PR (11/15), including 3 patients who had normalization of AFP, and four patients had CR (4/15). The levels of soluble VEGF and bFGF in sera of the control subjects varied from ND (not detected), to 300 pg/mL (with a mean value 140.75 pg/mL) for VEGF, and between 1.5 to 4.50 pg/mL (with a mean of 2.99 pg/mL) for bFGF (Table 1). In Group 1: statistical analysis of the readings of the patients indicated that, the sera levels of both angiogenic factors were reduced significantly (p < 0.05) after treatment as shown in table 1.
Profile of VEGF and bFGF in the different groups of patients.
| Group | No. of Patients | VEGF (pg/mL) Mean ± SD | bFGF (pg/mL) Mean ± SD | ||
|---|---|---|---|---|---|
| First | Last | First | Last | ||
| 1 | 15 | *684.8 ± 109.14 | 276.8 ± 137.6 | *23.1 ± 6.74 | *11.5 ± 5.15 |
| 2 | 19 | *618.47 ±184.34 | *1008.27 ± 203.8 | *26.97 ± 9.04 | *41.47 ± 4.58 |
| Control | 10 | 140.57 ±97.86 | 2.99 ± 0.96 |
* Statistically significant higher than control at p < 0.05.
In Group 2: before treatment, VEGF and bFGF concentrations were significantly higher than the control healthy subjects. A continuous increase in their levels was observed during the follow up duration (3 months) as shown in table 1. Angiogenic factors levels were measured every month during the time-course of the study (data not shown).
A liver biopsy was obtained from 15 patients of group1 and 19 patients of group 2 were analyzed for cytosolic level of PGE2 and VEGF before and after 3 months of treatment follow up in the tumor tissue. For group 1, treated with selective COX-2 inhibitor celecoxib, the median cytosolic level of PGE2 at time of diagnosis (zero time of treatment) was 0.735 ng/ mg total protein (range 0.34-1.2 ng/mg total protein) and VEGF median level was 97.9 ng/mg total protein (range 44-179.2 ng/mg total protein). The median cytosolic level of PGE2 in tumor samples, after 3 months of treatment with celecoxib, was 0.0665 ng/mg total protein (range 0.032-0.21 ng/mg total protein, (Figure 1) and VEGF median level was 18.7 ng/mg total protein (range 6.21-40.6 ng/mg total protein) (Figure 2). For group 2, treated with 5-FU only, at time of diagnosis the median cytosolic PGE2 concentration in the tumors was 0.795 ng/mg total protein (range 0.34-1.74 ng/mg total protein) and the median cytosolic VEGF concentration in the tumors was 79.9 ng/mg total protein (range 43.2-183.4 ng/mg total protein). The median cytosolic PGE2 level measured in tumor tissue after 3 months of treatment with 5-FU only, group 2, was 1.25 ng/mg total protein (range 0.75-2.7 ng/mg total protein) and the median cytosolic VEGF level was 151.15 ng/ mg total protein (range 66.21-290 ng/mg total protein) (Figures 1 and 2). The noncancerous adjacent tissues and their association with clinicopathologi-cal parameters have been reported in a previous pu-blication.13,14
 was observed in the samples of patients received celecoxib (group 1) after 3 months of treatment. On the contrary, there was a significant (p < 0.05) increase in tissue PGE2 level in the samples of patients treated with chemotherapy alone (group 2).")
Tissue expression of PGE2 in the two groups of HCC patients. PGE2 tissue level is elevated in all HCC patients before treatment. A significant reduction (p < 0.05) was observed in the samples of patients received celecoxib (group 1) after 3 months of treatment. On the contrary, there was a significant (p < 0.05) increase in tissue PGE2 level in the samples of patients treated with chemotherapy alone (group 2).
, parallel to the reduction of PGE2 level, was observed in the samples of patients received celecoxib (group 1) after 3 months of treatment. On the contrary, there was a significant (p < 0.05) increase in tissue VEGF level in the samples of patients treated with chemotherapy alone (group 2).")
Tissue expression of VEGF in the two groups of HCC patients. VEGF tissue level is elevated in all HCC patients before treatment. A significant reduction (p < 0.05), parallel to the reduction of PGE2 level, was observed in the samples of patients received celecoxib (group 1) after 3 months of treatment. On the contrary, there was a significant (p < 0.05) increase in tissue VEGF level in the samples of patients treated with chemotherapy alone (group 2).
Cytosolic fractions of liver biopsies were used for western blotting analysis of VEGF during the time-course. The results profile is consistent with the other data; an elevation of VEGF level was observed in all patients at time of diagnosis as shown in figure 3. A reduction of VEGF protein level after co-treatment with COX-2 inhibitor celecoxib was observed in the samples of patients of group 2 (Figure 3A). A continuous elevation was observed in the samples of patients treated with chemotherapy alone (Figure 3B).
 protein expression in HCC patients of groups 1 and 2. Samples of different patients from both groups were used for western blotting Assay (25μg protein). A. VEGF was high in all patients at zero time, and a significant reduction in VEGF level was observed after treatment with combined therapy, 5-FU and COX-2 inhibitor, celecoxib, for 3 months (group 1). B. VEGF level was high in three different patients before treatment, a significant elevation was observed in all patients treated with 5-FU alone (group 2). Beta actin was used as a loading control.")
Vascular endothelial growth factor (VEGF) protein expression in HCC patients of groups 1 and 2. Samples of different patients from both groups were used for western blotting Assay (25μg protein). A. VEGF was high in all patients at zero time, and a significant reduction in VEGF level was observed after treatment with combined therapy, 5-FU and COX-2 inhibitor, celecoxib, for 3 months (group 1). B. VEGF level was high in three different patients before treatment, a significant elevation was observed in all patients treated with 5-FU alone (group 2). Beta actin was used as a loading control.
A cytosolic fraction prepared from tumor biopsies was analyzed by western blotting to confirm that a group of 15 HCC patients have markedly higher expression of P-glycoprotein and COX-2 compared with parental cells (Figure 4A). To evaluate whether COX-2 activity induced the expression of P-glycopro-tein in these patients, we determined whether the selective inhibition of COX-2 activity by celecoxib modified P-glycoprotein and/or COX-2 expression after 3 months of treatment with combination of 5-FU, Endoxan and celecoxib. Western blot analysis clearly showed that the P-glycoprotein and COX-2 expression were reduced in the cytosolic fraction of all patients treated for 3 months with combination of 5-FU, Endoxan and celecoxib (Figure 4B).
.A. Western blot analysis of COX-2 and P-glycoprotein expression in the cytosolic fraction before treatment. B. Western blot analysis of COX-2 and P-glycoprotein expression after co-treatment with celecoxib for 3 months. Reduction of COX-2 and P-glycoprotein levels were observed concomitant with caspase-3 activation. β-actin was used in all western blot analyses as a protein loading control.")
Effect of co-treatment of HCC patients with celecoxib on COX-2 and P-glycoprotein expression. Two separate experiments were done for all patients of group 1 and one representative blot is shown (3 lanes of 3 different patients).A. Western blot analysis of COX-2 and P-glycoprotein expression in the cytosolic fraction before treatment. B. Western blot analysis of COX-2 and P-glycoprotein expression after co-treatment with celecoxib for 3 months. Reduction of COX-2 and P-glycoprotein levels were observed concomitant with caspase-3 activation. β-actin was used in all western blot analyses as a protein loading control.
The expression of caspase-3 was evaluated by Western blot analysis. Treatment with celecoxib was shown to activate caspase-3 in the COX-2 and p-glycoprotein positive tumor samples of HCC patients after 3 months of treatment. Western blot analysis of GPC3 expression in the cytosolic fraction before treatment and after 6 months of treatment indicated reduction of GPC3 protein level. β-Actin was used in all western blot analyses as a protein loading control (Figure 5).

Inhibition of COX-2 expression was confirmed by RT-PCR analysis of COX-2 mRNA level before and after treatment of patients of group 1. A significant reduction in COX-2 mRNA level was detected after 6 months of treatment with combined therapy (Figure 6).
 for 6 months compared to the samples before treatment.")
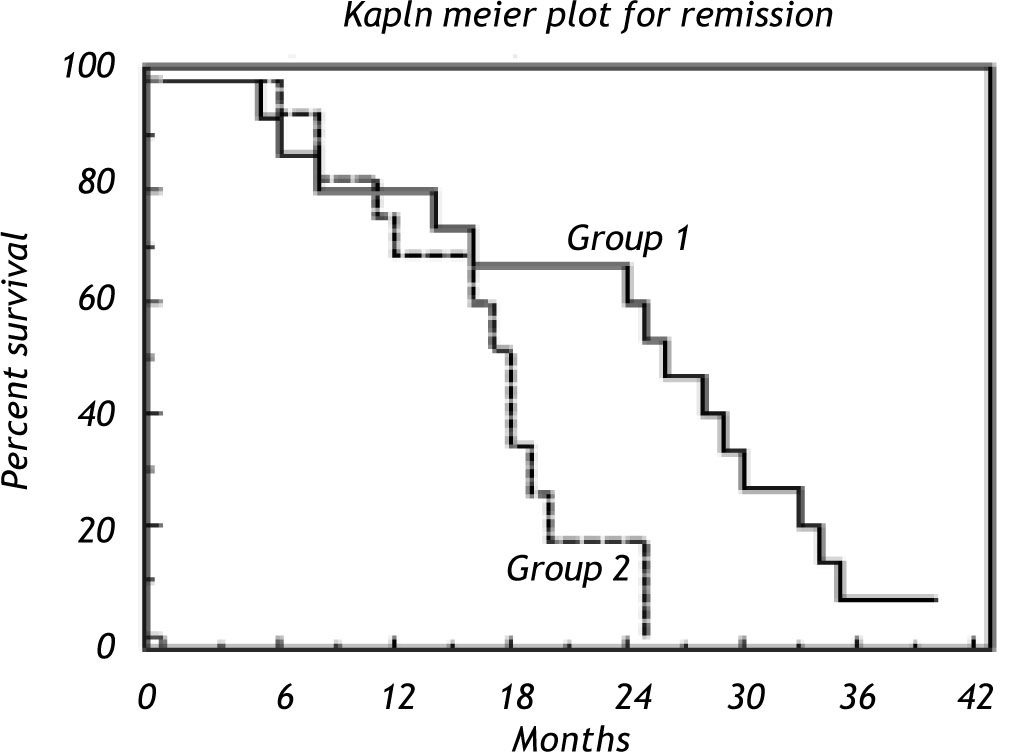
Evaluation of overall survival (OS) and progression-free survival (PFS) results indicated that 15 patients in group I with a median survival 26 with 0.3179 hazard ratio at (95% confidence intervals and the median OS was 26. The median survival for group II is 18. Logrank test shows a statistical significance with P = 0.0232 (Figure 7).
Discussion
Recent studies have demonstrated that expression of COX-2 is positively correlated with the P-glycopro-tein (p170, P-gp) encoded by human MDR1 gene, suggesting a potential of COX-2 specific inhibitors in regulation of cytotoxicity of anticancer agents. Cele-coxib is one of the specific inhibitors of COX-2 and has been widely used in clinic. However, its function in the response of cancer cells to anticancer drugs and the related mechanism are still waiting to be in-vestigated.19 The MDR phenotype is characterized by the overexpression of P-glycoprotein in plasma membrane that works as a pump to extrude anticancer drugs from cells. However, in addition to the overex-pression of P-glycoprotein, the MDR phenotype is associated with other modifications of cell biology that make cancer cells more resistant to many other mechanisms of cell damage.20,21 It was previously shown that the development of the MDR phenotype is associated with the constitutive expression of COX-2 in hepatocellular carcinoma cell lines.22 P-glycoprotein can regulate the mitochondrion-dependent apoptotic pathway, conferring resistance to a caspase-depen-dent mechanism of apoptosis induced by antineoplas-tic agents.14,23 The antitumor effect of COX-2 inhibitors has been described in recent years although the molecular mechanisms involved remain elusive.24,25 NF-κB is frequently abnormally present in clinical HCC;26,27 its persistent and inappropriate activation in hepatocytes is implicated in tumor initiation and progression following events like viral infection (HBV or HCV), exposure to carcinogens, growth factor stimulation (TGF-α, HGF/SF and TGF-β) and inflammation, as recently reviewed by Arsura and Cavin.28 The present study was conducted to decipher, in vivo, the role of Celecoxib on the expression of cyclooxygenase (COX)-2 and nuclear factor kappa B (NF-κB)-p65 during hepatocarcinoge-nesis in human subjects. Reverse transcriptase poly-merase chain reaction (RT-PCR) analysis revealed that occurrence of HCC in humans caused inflammation of the liver due to up-regulation of NF-κB and COX-2. RT-PCR and immunoblot analysis also revealed that the oral supplementation of Celecoxib (400 mg/day) to hepatocellular carcinoma patients down-regulated the expression of COX-2 and NF-κB-p65, thereby preventing inflammation and angiogenesis mediated hepatocellular carcinogenesis.
In the present study, we explored the effect of ce-lecoxib on COX-2 and P-glycoprotein expression and on factors involved in the mitochondrial apoptosis pathway, in hepatocellular carcinoma patients. The relationship between COX-2 and P-glycoprotein suggests that celecoxib, a specific COX-2 activity inhibitor, may improve results of chemotherapy by increasing the sensitivity of tumor cells to antican-cer drugs. Our data show that the combination therapy containing celecoxib caused marked regression of tumor size when HCC patients were treated with 400 mg/day celecoxib. We suggest that release of cytochrome c into cytosol initiates the activation of caspase-3 and stimulates apoptosis. Our results are in agreement with other studies that have shown a COX-2-mediated regulation of P-glycoprotein ex-pression.29
It is generally accepted that solid tumor growth and metastasis are dependent on the acquisition of an adequate blood supply.30 Pharmacological targeting of microvasculature in patients with cancer represents an attractive therapeutic approach because inhibition of angiogenesis has been shown to prevent growth,31 and induce regression of experimental solid tumors.
To investigate the angiogenic pathway that may be involved in COX-2 activity, we evaluated the expression of VEGF & bFGF in all HCC patients included in our study with different treatment regimens. Table 1 showed the serum profile of VEGF & bFGF levels in the HCC patients. In support of our results, it has been shown that VEGF plays an important role in determining the biological aggressiveness of neoplastic cells and thus, their metasta-tic potential.32
Concerning the patients treated with combination of chemotherapy and selective COX-2 inhibitor cele-coxib the serum levels of glypican-3, VEGF and bFGF were significantly higher than that of the control subjects before treatment. A significant reduction in both VEGF and bFGF serum levels was observed in all patients receiving the selective inhibitor in combination with chemotherapy as a first response to the treatment. Our study showed that tissue and serum GPC3 levels were significantly elevated in all 15 patients with MDR-positive-HCC, and in 81% of other patients but were rarely detectable in patients with non-malignant chronic liver disease and healthy controls. This is in agreement with Tangkijvanich, et al., study.33 Serum GPC3 was superior to AFP in detecting small HCC and a combination of serum GPC3 and AFP yielded an improved sensitivity for detecting small HCC to 83%. Interestingly, in most cases, elevated GPC3 values did not correlate with elevated AFP values. As a consequence, the serological level of at least one of the two markers was elevated in a large majority of HCC patients.
In conclusion, the induction of MDR1 expression by PGE2 and its downregulation by Celecoxib suggests that the enhanced sensitivity of HCC tumor to 5-FU by Celecoxib is mediated by the down regulation of MDR1 expression, through COX-2-depen-dent mechanism. Further studies revealed the involvement of AP-1 in the Celecoxib-induced do-wnregulation of MDR1 expression.34 The present study thus demonstrates the efficacy of COX-2 intervention in overcoming the drug resistance in HCC tissues. Therefore, these data might provide new therapeutic modalities for HCC prevention and treatment by enhancing the sensitivity of HCC cells to anticancer drug via selective pharmacologic inhibition of COX-2.
AcknowledgmentsWe gratefully acknowledge Dr. Ahmed Kaseb, Assistant Professor, Department of Gastrointestinal Medical Oncology, The University of Texas M. D. Anderson Cancer Center USA for intellectual inputs and critical advice on the manuscript, Dr. Ahmad Youssry, Mabarret Al-Assafra for technical assistance and lab facility, and by the National Al-Amereya Pharmaceutical Company (Part of Pharco Companies), Alexandria, Egypt for providing us with the drug.
This work was supported by Faculty of Science and Faculty of Medicine, Alexandria University and by personal funds and communication.



