




Sepsis is a potentially life-threatening systemic inflammatory response syndrome caused by the spread of pathogens (such as bacteria or viruses) and their toxins in the blood, and can result in septic shock and multiple organ dysfunction syndrome in severe cases [1]. As an important metabolic organ in the body, the liver plays a central role in metabolic and immune homeostasis, and is closely associated with immune homeostasis imbalance [2]. Clinical evidence shows that liver dysfunction is an important reason for poor prognosis in patients with sepsis-induced multiple organ failure [3]. During sepsis, the liver undergoes inflammation, oxidative stress damage, and cell death [4]. Therefore, it is crucial to identify protective mechanisms against sepsis-induced liver injury to restore liver function and reduce liver mortality.
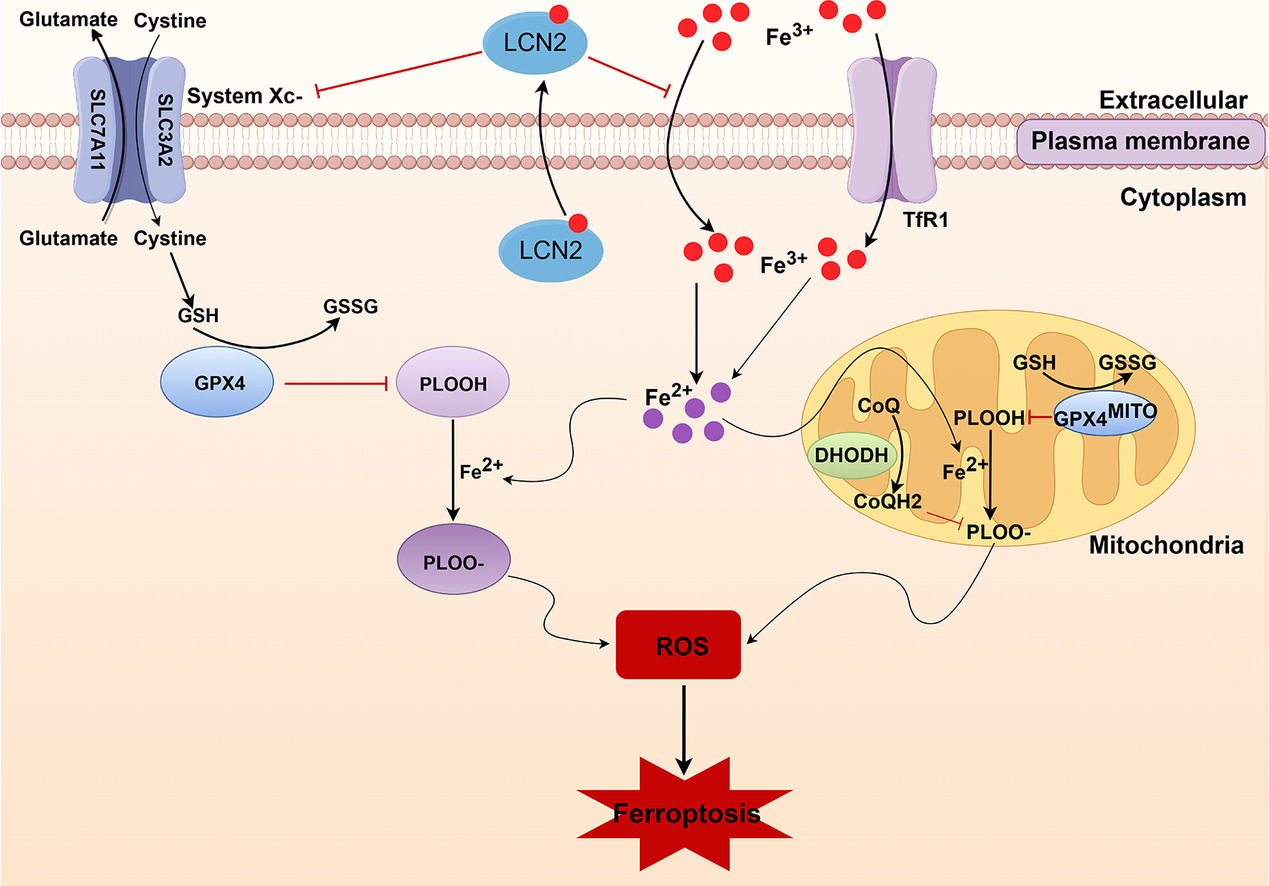
Ferroptosis, an iron-dependent regulatory cell death mode, is mainly induced by free iron and reactive oxygen species (ROS) [5] [5], leading to the inactivation of the antioxidant defense system in cells. Transferrin receptor 1 (TfR1) is a major regulator of iron uptake in cells and serves as a carrier protein for transferrin (Tf) [6]. Increased TfR1 expression is considered to be an important factor in the dysregulation of cellular iron homeostasis [7]. The cyst(e)ine/glutathione (GSH)/glutathione peroxidase 4 (GPX4) axis is the main cellular pathway that mediates ferroptosis [8]. GPX4 is a key enzyme required to clear lipid ROS from the body, and its activity depends on the activation of cysteine-glutamate anti transporter xCT [9]. Therefore, xCT protein levels reflect the occurrence of ferroptosis. Wang et al. and our group previously reported that GPX4 protein expression is decreased during sepsis-induced liver injury. However, changes in the other iron metabolism-related proteins have not been determined [10]. Dihydroorotate dehydrogenase (DHODH) functions as an antioxidant to inhibit ferroptosis in a GSH-independent manner. Inhibition of DHODH promotes ferroptosis by increasing lipid peroxidation in the mitochondria, providing a new strategy for cancer treatment [11]. Recently, our group reported that GPX4 and ferroptosis suppressor protein 1 (FSP1)-mediated ferroptosis are involved in the development of septic liver injury [12]; however, whether the mitochondrial DHODH-mediated ferroptosis inhibition mechanism is involved in the pathogenesis of septic liver injury remains unclear.
Mitochondrial DHODH, a rate-limiting enzyme of de novo biosynthesis of pyrimidine [13], plays a pivotal role in cellular metabolic balances. During ferroptosis, DHODH reduces ubiquinone (also known as coenzyme Q, CoQ) to dihydroubiquinone (CoQH2) in the mitochondrial inner membrane and inhibits ferroptosis by preventing lipid peroxidation, providing a potentially new target for the treatment of breast, colorectal, lung, and other cancers [14,15]. Many studies have shown that ferroptosis is involved in cancer, neurodegenerative diseases, myocardial injury, acute lung injury, and other diseases [16,17], including septic liver injury. Wang et al. reported that the expressions of liver ferroptosis-related proteins GPX4, solute carrier family 7 member 11 (SLA7A11), and ferritin heavy chain polypeptide 1 (FTH1) was significantly reduced in mice with septic liver injury [18]. However, the changes and mechanisms of other key molecules and pathways in ferroptosis, and whether DHODH inhibition mediates ferroptosis remain unclear.
Lipocalin-2 (LCN2) is a secreted glycoprotein that induces a variety of chemokines to participate in inflammatory responses, and plays a key role in various biological processes [10]. LCN2 serves as a biomarker for the severity of renal injury and hepatic ischemia-reperfusion injury [19,20]. Clinical studies have revealed that serum LCN2 (also named NGAL) levels are markedly elevated in various types of liver diseases [21], and LCN2 is a predictor for mortality and bacterial infection in patients with liver cirrhosis, suggesting that LCN2 is associated with bacterial infection in liver cirrhosis [22]. LCN2 is upregulated under various cellular stress conditions, and is an important regulator of iron metabolism, oxidative stress, and inflammation in mammals [23,24]. In acute respiratory distress syndrome, LCN2 knockdown reduced iron content and Tf levels in and out of cells, increased FTH1 and GPX4 levels, and decreased ROS levels, suggesting that LCN2 inhibition may reduce inflammation and oxidative stress by reducing ferroptosis [25]. Our previous showed that LCN2 protein levels were increased in mice with septic myocardial injury [26], however, the changes of LCN2 in septic liver injury, and whether LCN2 is involved in the occurrence of ferroptosis have not been investigated.
Therefore, we used the cecal ligation and puncture (CLP) method to induce sepsis in mice to observe the cellular changes during liver injury. In addition, we used the ferroptosis-specific inhibitor ferrostatin-1 (Fer-1) and iron chelator dexrazoxane (DXZ) to analyze the roles of ferroptosis and LCN2 in septic liver injury. Furthermore, we knocked down LCN2 expression through AAV9-shLCN2 to elucidate its possible regulatory role. The objective of the study was to provide novel insights into the mechanisms underlying septic liver injury.
2Materials and methods2.1AnimalsMale C57BL/6 mice (6–8 weeks old, body weight 20–25 g) were purchased from Henan SkBys Biotechnology Co., LTD (Henan, China). The animals were maintained in a clean environment at a constant temperature (22−25 °C) with a 12-h dark/light cycle with free access to food and water.
To examine the effect of ferroptosis in sepsis-induced liver injury, we randomly assigned the mice to six groups (10 mice/group) as follows: Sham, CLP, CLP + DMSO (lysis medium), iron overload (iron-dextran, mice were injected intraperitoneally with iron-dextran at 1000 mg/kg [27]), CLP + ferroptosis inhibitor Fer-1(mice were injected intraperitoneally with Fer-1 [5 mg/kg] 2 h before CLP [28]), CLP + iron chelator DXZ (mice were injected intraperitoneally with DXZ [50 mg/kg] 2 h before CLP [29]). To examine the mechanisms underlying LCN2 silencing-mediated protective effects against sepsis liver injury, mice were assigned to four groups (10 mice/group) as follows: Sham, CLP, CLP + NCRNA (negative control, transfected with the empty sequence), and CLP + ShRNA-LCN2 (knockdown LCN2 expression).
2.2CLP mouse modelA mouse model of sepsis was established using CLP. All animals were fasted for 12 h prior to surgery. Mice were continuously anesthetized with isoflurane mixed with pure oxygen using an inhalation anesthesia machine. The abdomen was disinfected and a longitudinal incision of 1 cm was made at the midline of the abdomen. The peritoneum was cut open, and the cecum was carefully separated to avoid vascular injury. After the distal 1/3 of the cecum was ligated, both sides of the cecum were punctured twice with an 18-G needle and a small amount of intestinal content was squeezed out. The cecum was returned to the abdominal cavity and the abdomen was sutured with sterile sutures after disinfection. The mice were resuscitated by subcutaneous injection of prewarmed normal saline (37 °C, 1 mL/20 g). Mice in the Sham group underwent the same procedure as described above, except that the cecum was not ligated or perforated. After 24 h, detection and analyses were performed [6].
2.3Adeno-associated virus(AAV) infection of LCN2To knockdown LCN2 in vivo, we transduced mice with AAV serotype 9 encoding a green fluorescent protein reporter together with either short hairpin RNAs targeting LCN2 (shRNA-LCN2) or an empty vector (NCRNA) [30] in the liver. ShRNA sequences targeting mouse LCN2 were cloned into AAV by Genechem Co., Ltd. (Shanghai, China). Three weeks prior to the establishment of the CLP model, mice were injected with AAV carrying shRNA targeting LCN2 or vehicle alone (1 × 1011 viral particles/mouse) via tail vein once.
2.4Liver function analysesBlood samples were collected, incubated at room temperature for 2 h, and then centrifuged at 3000 rpm at 4 °C for 15 min to isolate the serum. Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) levels were measured using the corresponding commercial Enzymatic Assay Kits (Cat# C0092-1 and Cat# C010-2-2, respectively; Jiancheng, Nanjing, China).
2.5Measurement of lipid peroxidation indicators in liver tissueLiver tissue samples were collected from the mice, and tissue homogenates were prepared according to the instructions in the corresponding assay kit. The lipid peroxidation products malondialdehyde (MDA; Cat# M496, Dojindo, Shanghai, China) and superoxide dismutase (SOD; Cat# A001-3, Jiancheng, Nanjing, China) in the liver tissue were measured using the corresponding assay kits.
2.6Hematoxylin and eosin (H&E) stainingFresh liver tissue samples were collected, washed in PBS, fixed with 4 % paraformaldehyde for 24 h, dehydrated in ethanol gradient, and embedded in paraffin. Sections of the fixed liver tissue were stained with H&E. Changes in liver tissue structure were analyzed under a light microscope (Nikon Eclipse E100).
2.7Transmission electron microscopy (TEM)Liver tissue samples were fixed with 2.5 % glutaraldehyde in phosphoric acid buffer for 2 h, followed by post-fixation with 1 % osmium acid for 2 h. The tissue samples were sectioned and stained. The sections were dehydrated in ethanol, embedded in acetone, polymerized and stained. Ultrastructural images of the liver were visualized by TEM (HT7800, Hitachi).
2.8Detection of ROS levels in liver tissueROS levels in the liver tissues were measured using a dihydroethidium (DHE) fluorescence probe. Fresh liver tissue samples were collected, embedded in OCT, frozen at −80 °C, and cut into 10 µm thick sections. The tissue sections were stained with 10 µmol/mL DHE (Cat# S0063, Beyotime, Shanghai, China) and 5 µmol/mL diaminophenyl indole staining (DAPI, Cat# S0063, Beyotime) for 30 min at 37 °C protected from light. The fluorescent images were captured using the Live Cell Imaging Workstation (Cat # TY2014009656, Zeiss). The results were analyzed using the Zen image analysis software (Zeiss).
2.9Western blot analysisTotal proteins were extracted from liver tissues using lysis buffer containing protease inhibitors. Protein concentration in each sample was determined using the bicinconinic assay method. Proteins were denatured by adding 5 × protein loading buffer, and then separated by 10 % sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) (Cat# PG112, Epizyme Biomedical Technology Co., Ltd, Shanghai, China). Following electrophoresis, the proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (Cat# IPVH00010, Millipore Sigma, USA). The membrane was blocked with 5 % skim milk for 2 h, washed with tris-buffered saline (TBS)/Tween (TBST), and then incubated overnight with primary antibodies against LCN2 (1:1000, Cat# AF1857, R&D system, USA), DHODH (1:6000, Cat# 14877-1-AP, Proteintech, China), xCT (1:5000, Cat# ab175186, Abcam, USA), TfR1 (1:1000, Cat# 13-6800, ThermoFisher, USA), GPX4 (1:3000, Cat# ab125066, Abcam, USA), GADPH (1:6000, Cat# 60004-1-lg, Proteintech, China). The next day, the membranes were washed three times with TBST, incubated with goat anti-rabbit (Cat# BL003A, Biosharp, China), goat anti-mouse (Cat# BL001A, Biosharp, China), or rabbit anti-goat (Cat# BA1060, Boster Bio, China) secondary antibodies for 2 h, and washed four times with TBST. The protein bands were detected using chemiluminescence (Cat# FluorChem M; ProteinSimple, USA). Protein levels in the samples were analyzed using the ImageJ software.
2.10Statistical analysisAll data are expressed as the mean ± standard deviation. The groups were compared using one-way analysis of variance and Tukey's test. Statistical significance was set at P < 0.05. All results were plotted using the GraphPad Prism 9 software.
2.11Ethical statementsThis study was approved by the Animal Ethics Committee of BengBu Medical University (Ethics Number: [2023] No. 523). Care and handling of the animals were carried out in strict accordance with the Regulations on the Management of Laboratory Animals.
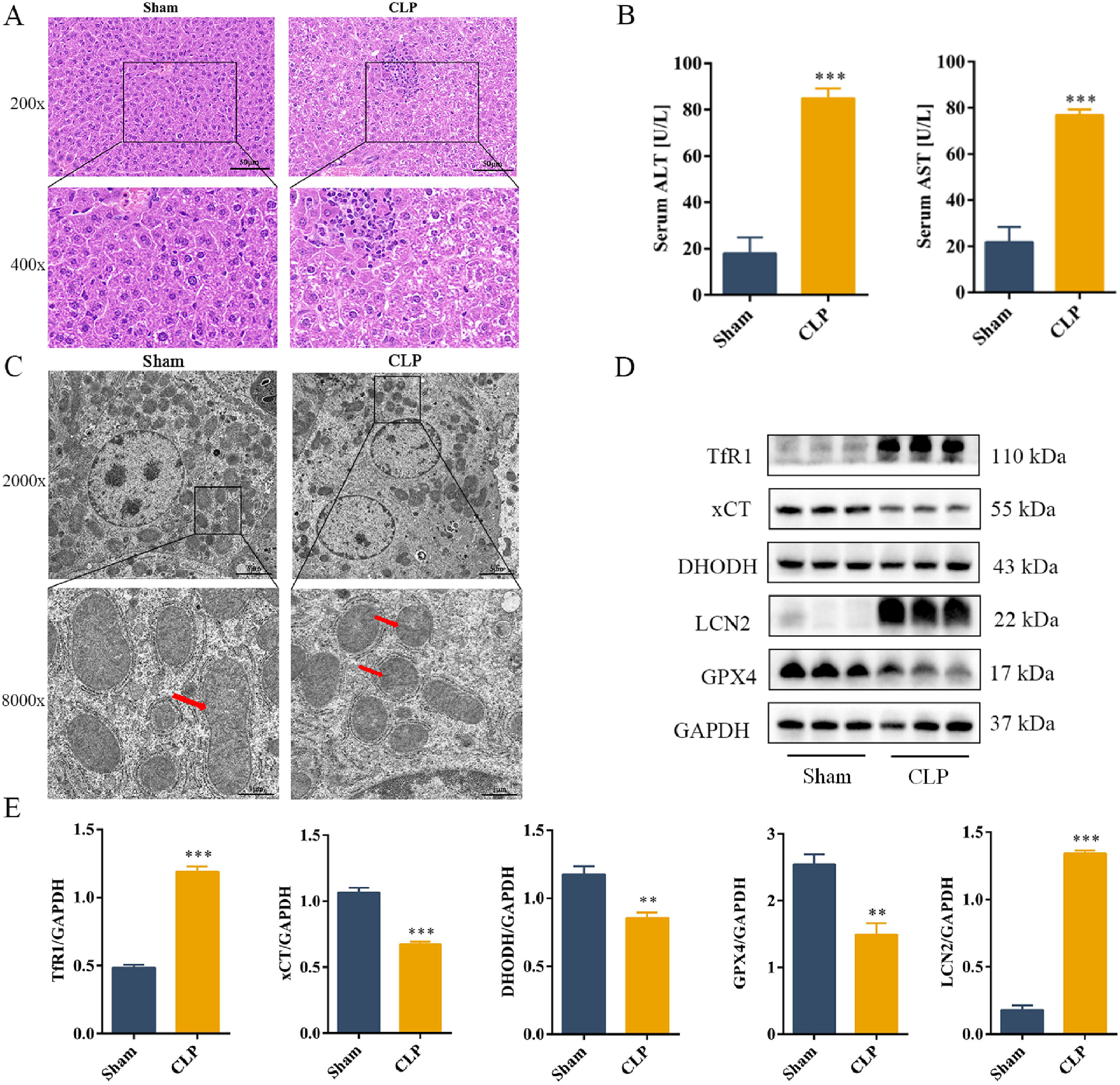
3Results3.1CLP induced liver damage in mouse by activating ferroptosis in the liver tissueH&E staining showed that the hepatocytes in the Sham group were arranged neatly, with normal hepatic lobule structure, and no significant change in their morphology. Compared to the Sham group, the CLP group showed severe structural damage, erythrocyte exudation, large amount of spot necrosis, and inflammatory cell infiltration (Fig. 1A) Serum levels of ALT and AST were increased in the CLP group (Fig. 1B). TEM was used to assess mitochondrial morphology to determine whether the aggravated liver injury was mediated by ferroptosis. Sepsis induced significant morphological changes in the mitochondria, including smaller mitochondria and reduced cristae (Fig. 1C). Analysis of the levels of key ferroptosis-related protein showed that the expression levels of TfR1 and LCN2 increased, whereas those of the anti-ferroptotic proteins GPX4, xCT, and DHODH decreased during sepsis (Fig. 1D and E). In summary, our results suggested that LCN2 is involved in ferroptosis during sepsis-induced liver injury in mice.

CLP induces liver damage in mouse by activating ferroptosis in the liver tissue.
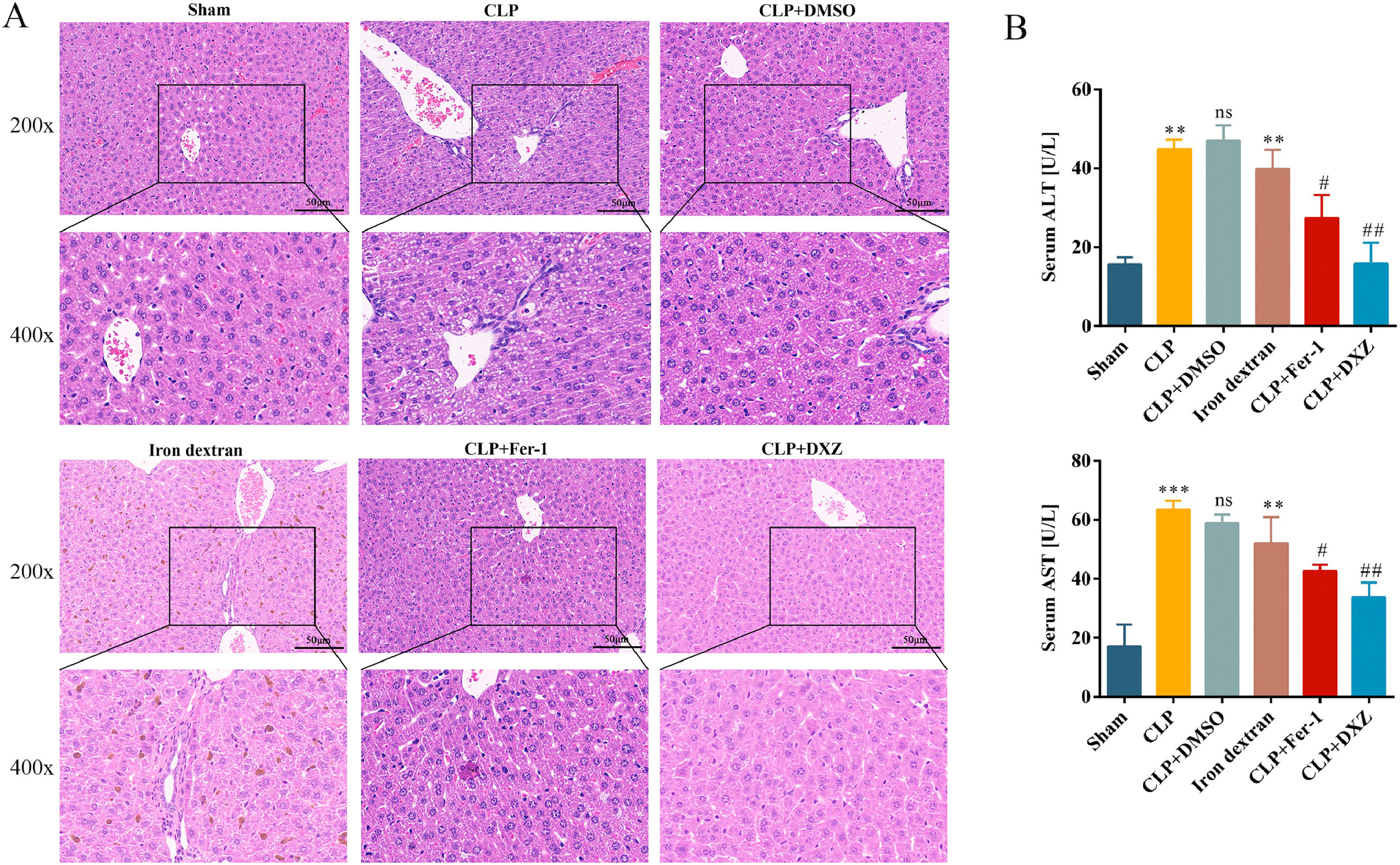
Next, we examined the effect of ferroptosis inhibition on liver damage caused by sepsis. We used ferroptosis inhibitors and iron-dextran to confirm the role of ferroptosis in the development of CLP-induced liver injury. Fer-1 and DXZ were used to inhibit ferroptosis, whereas iron-dextran was used to induce ferroptosis. As shown in Fig. 2, the CLP and iron-dextran groups showed liver structural damage, hepatocyte necrosis, and inflammatory cell infiltration. However, after treatment with Fer-1 and DXZ, the extent of liver injury was obviously attenuated (Fig. 2A). Compared with the Sham group, the serum levels of ALT and AST were significantly increased in the CLP and iron-dextran groups, whereas after treatment with Fer-1 and DXZ, the serum levels of ALT and AST were decreased compared to those in the CLP group. No significant difference was noted in the CLP + DMSO group (Fig. 2B).

Ferroptosis inhibition alleviates liver damage caused by sepsis.
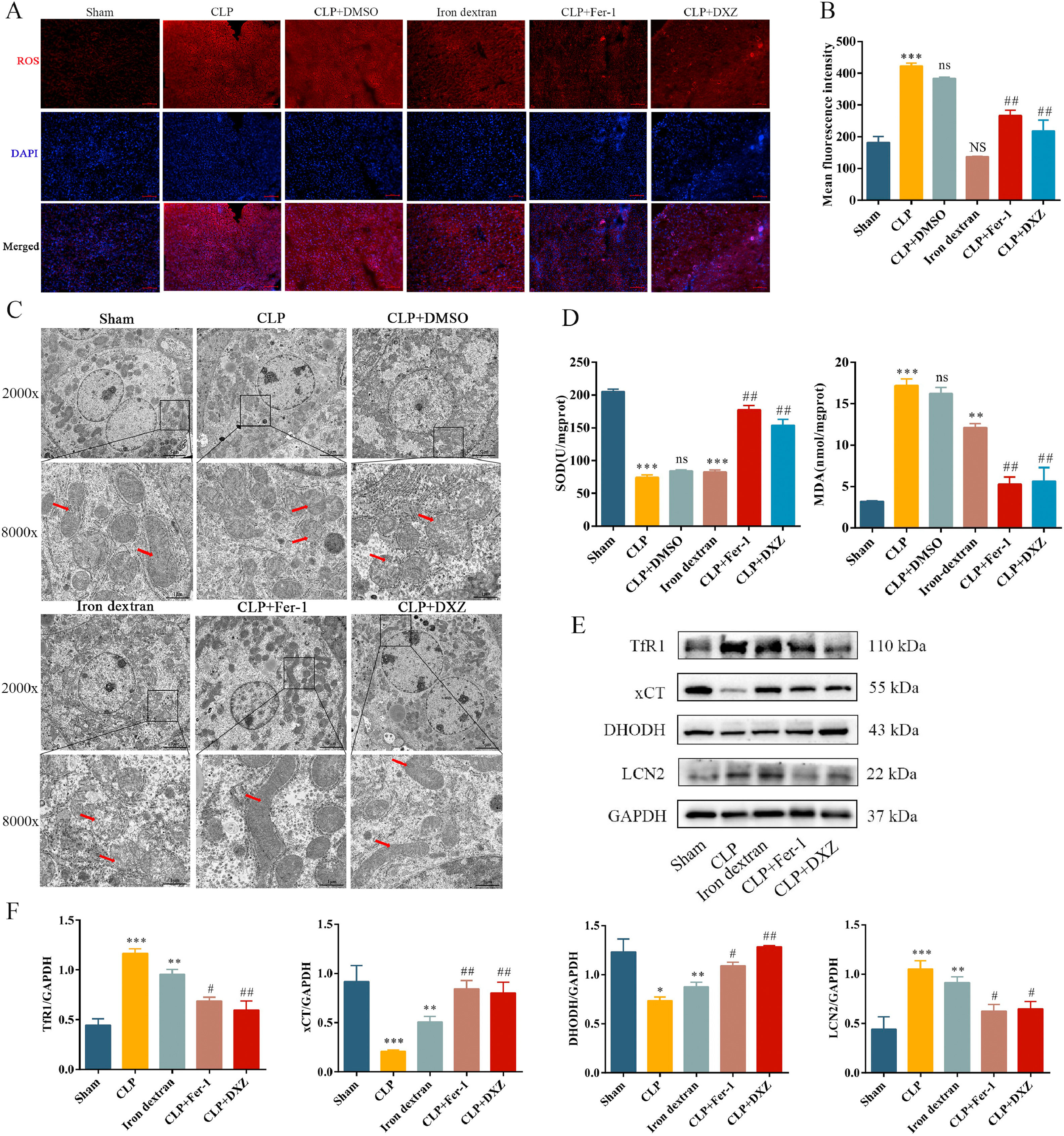
Sepsis-induced liver injury is often accompanied by oxidative stress dysfunction, which leads to accumulation of lipid ROS [31], and production of membrane lipid peroxides, and continued oxidative stress causes hepatocyte death [32]. Our results showed that CLP significantly increased ROS levels in the liver. After treatment with Fer-1 and DXZ, ROS (Fig. 3A, B) and MDA (Fig. 3D) levels decreased and SOD level (Fig. 3D) increased. Iron-dextran caused an increase in SOD and MDA levels, but ROS levels were not significantly different from those in the Sham group. TEM revealed that CLP and iron-dextran caused significant increase in aberrant mitochondria, including shrunken mitochondria, reduction/loss of mitochondria cristae, and rupture of mitochondrial outer membrane. However, following intervention with Fer-1 or DXZ, the mitochondrial morphology returned to almost normal (Fig. 3C). These results showed that the CLP group did not differ significantly from the CLP + DMSO group. Measurement of ferroptosis-related protein levels revealed that iron-dextran exacerbated liver ferroptosis, whereas Fer-1 and DXZ inhibited ferroptosis, with increased levels of TfR1 and LCN2 proteins, and decreased levels of xCT and DHODH proteins (Fig. 3E and F). These results indicate that ferroptosis occurs during sepsis and plays a destructive role in sepsis-induced liver injury.

Inhibition of ferroptosis regulates sepsis-induced liver injury by reducing lipid peroxidation and iron overload.
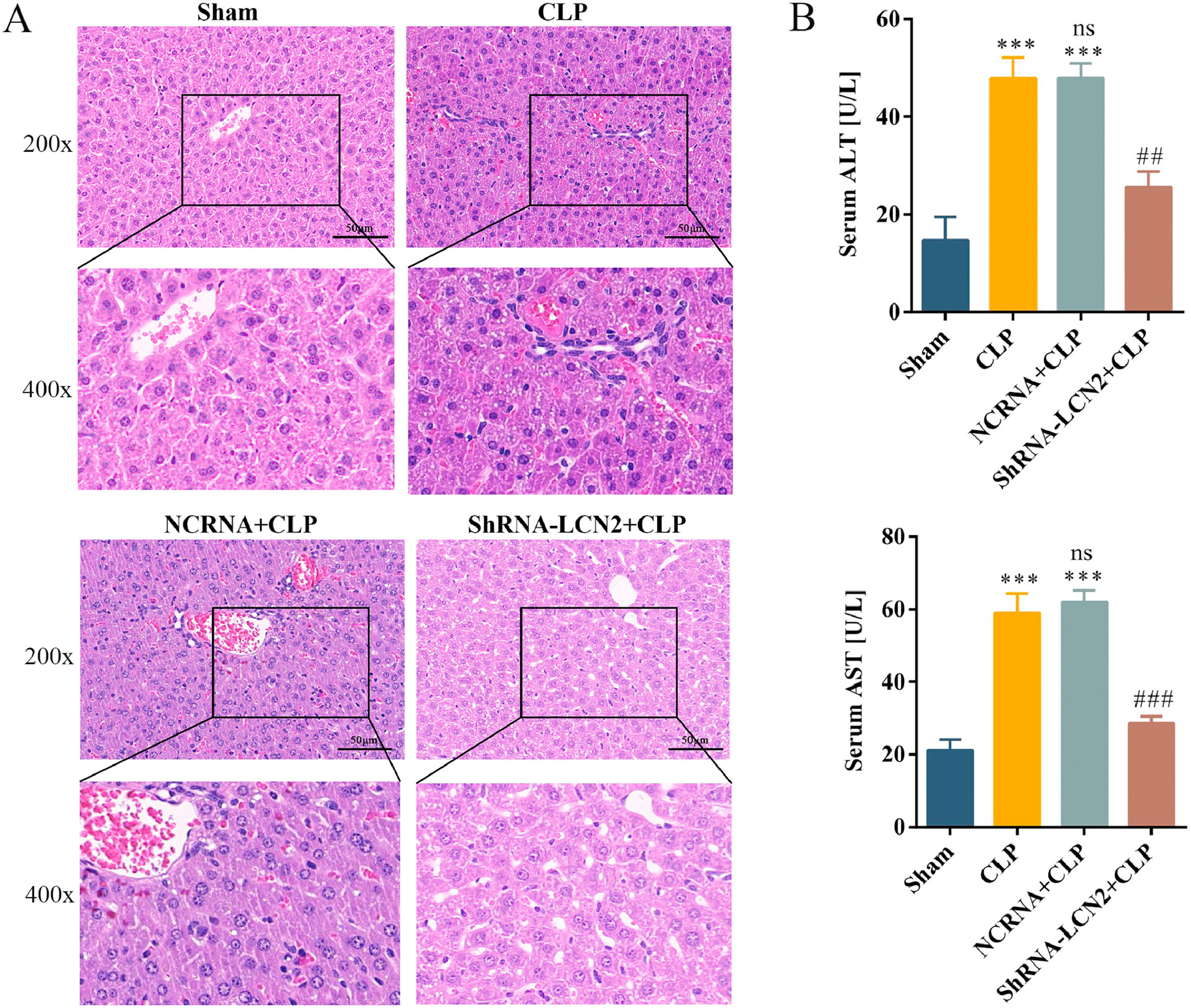
To examine the role of LCN2 in septic liver injury, mice were injected with AAV9-shLCN2 to knockdown LCN2 expression. Mice in the CLP group showed severe structural damage and increased serum ALT and AST levels, whereas mice in the shRNA-LCN2 + CLP group showed improved liver dysfunction when compared with mice in the NCRNA + CLP group. H&E staining results showed more intact liver lobules, lower inflammatory infiltration, erythrocyte exudation, and decreased levels of ALT and AST in the shRNA-LCN2 + CLP group (Fig. 4).

LCN2 knockdown attenuates liver damage caused by sepsis.
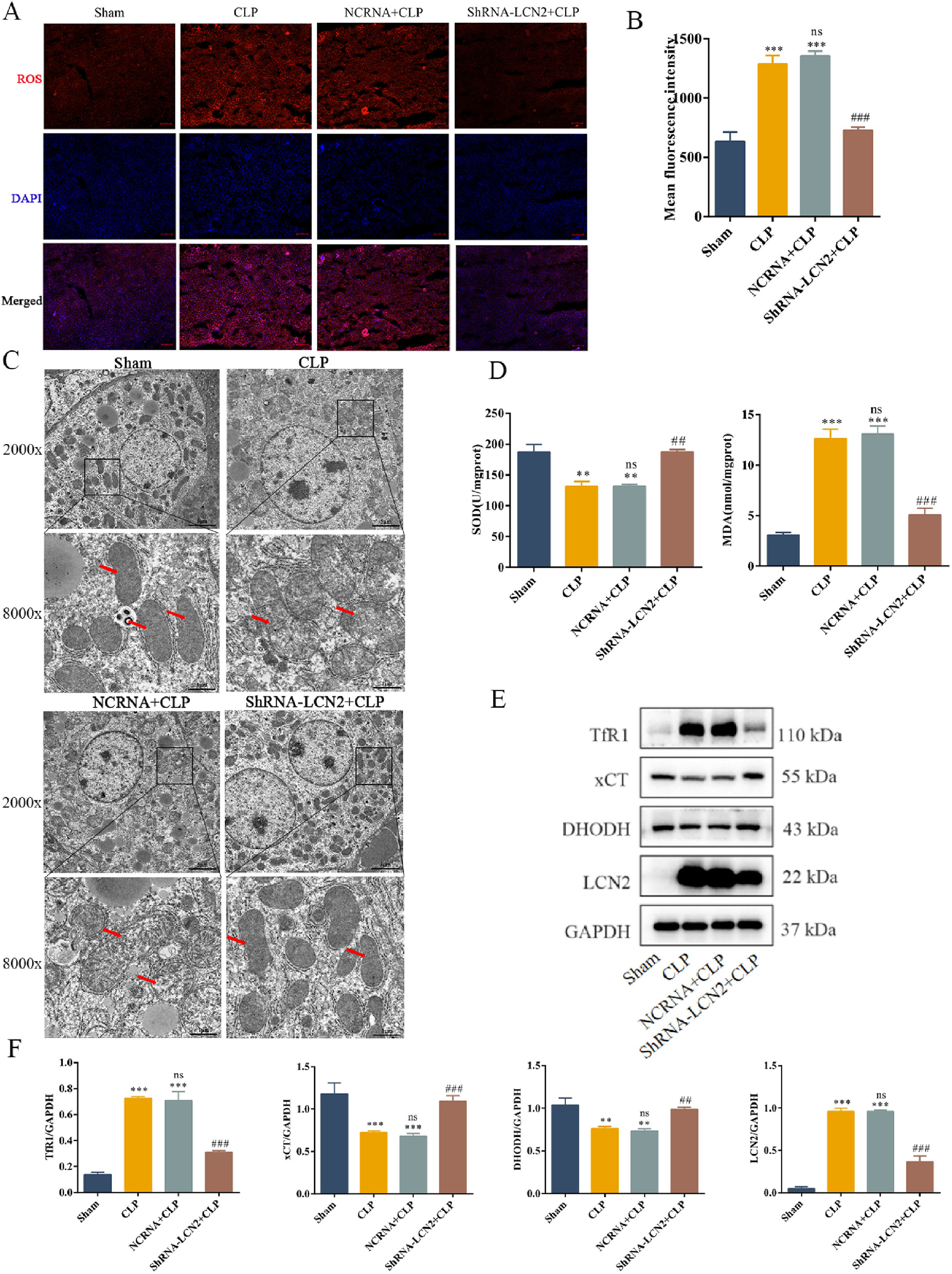
To determine whether LCN2 knockdown suppresses ferroptosis, we examined ferroptosis-associated proteins in AAV9-shLCN2-injected mice. Both serum ROS and liver MDA levels were markedly increased in the shRNA-LCN2 + CLP group compared with those in the NCRNA + CLP group (Fig. 5A and B). TEM showed smaller and disordered mitochondria, reduced mitochondrial cristae, and rupture of the mitochondrial outer membrane in the shRNA-LCN2 + CLP group (Fig. 5C). Furthermore, SOD levels were lower in the liver tissue in the shRNA-LCN2 + CLP group (Fig. 5D) when compared with that in the NCRNA + CLP group. TFR1 and LCN2 protein levels were increased in CLP mouse liver, but reduced in mice with LCN2 knockdown, whereas xCT and DHODH protein levels were decreased in CLP mouse liver, but increased in mice with LCN2 knockdown (Fig. 5D and E). These data indicated that LCN2 knockdown suppresses sepsis-induced lipid peroxidation and ferroptosis.

LCN2 knockdown protects mice against sepsis by inhibiting lipid peroxidation and ferroptosis.
The liver is an important organ in immune defense, and vulnerable to sepsis-induced damage. Clinical studies have shown sepsis-induced liver injury is closely associated with increased mortality. Excessive inflammatory response and oxidative stress damage are important causes of this complication [33]. In the present study, CLP was used to induce septic liver injury in mice. The results showed that sepsis caused severe pathological changes in the liver, manifesting as liver tissue edema and necrosis, significantly increased serum AST and ALT levels, increased ROS and MDA levels in the liver tissue, and decreased SOD activity, suggesting that sepsis causes oxidative stress and injury in the liver of the CLP mouse model.
Iron overload and ferroptosis are inseparable, excessive free iron catalyzes the Fenton reaction to produce a large number of oxygen free radicals, causing severe oxidative stress. Liver iron overload may lead to liver fibrosis, cirrhosis, and hepatocellular carcinoma by promoting hepatocyte death [34,35]. In the present study, we observed that TfR1 protein level increased, whereas that of xCT and DHODH proteins decreased in sepsis and iron overload-induced liver injury, indicating that ferroptosis is involved in sepsis and iron overload-induced liver injury through different ferroptosis pathways. Huang et al. used LPS/D-GalN to induce acute liver failure in mice, and found that the levels of GPX4 and xCT expressions were decreased, DHODH expression was also inhibited, and TfR expression was increased [36]. Their results are consistent with the results of the present study, suggesting that different ferroptosis inhibition pathways cooperate to protect the hepatocytes from injury. However, the specific underlying mechanisms require further investigations.
The ferroptosis inhibitor Fer-1 and mitochondrial permeable iron chelator DXZ inhibit lipid peroxidation and ferroptosis, and maintain normal mitochondrial function by scavenging oxygen free radicals and iron accumulated in the mitochondria [37,38]. To further analyze the role of ferroptosis in liver injury in septic mice, we used Fer-1 and DXZ intervention, and found that Fer-1 and DXZ reduced liver oxidative stress and lipid peroxidation, improved liver tissue injury and inflammatory response, decreased TfR1 protein expression, and increased xCT and DHODH protein expression in CLP mice. These results indicated that the inhibition of ferroptosis and chelation of iron ions alleviate lipid peroxidation and liver injury caused by sepsis.
Ferroptosis is closely associated with LCN2. As an iron transporter, LCN2 transport siderophore-bound iron to cells and increases the cytoplasmic labile iron pool (LIP) [39], leading to ferroptosis. Studies have shown that LCN2, a bacteriostatic factor, inhibits bacterial growth by binding to siderophores and preventing bacterial acquisition of iron, and thus plays a protective role in infectious and inflammatory bowel diseases [40]. However, LCN2 may induce ferroptosis by increasing the LIP in neurodegenerative diseases, kidney diseases and cancer [41]. LCN2 knockout reduced myocardial ferroptosis and improved cardiac function in mice with septic cardiomyopathy. Silencing LCN2 improved the viability [42] of cardiomyocytes under iron overload conditions by reducing mitochondrial dysfunction and apoptosis. In the present study, we observed that LCN2 protein expression was increased in the liver tissue of septic mice, suggesting that increased LCN2 expression may be an important cause of liver injury in septic mice. To further determine the role of LCN2 in liver injury during sepsis, mice were injected with AAV9-shLCN2 to knockdown LCN2. The results showed that LCN2 knockdown attenuated sepsis-induced liver damage, reduced oxidative stress, and increased the expression of anti-ferroptosis proteins xCT and DHODH, suggesting that LCN2 is associated with the occurrence of ferroptosis in septic liver injury, and that LCN2 knockdown reduces ferroptosis in the liver of septic mice. Considering the multiple functions of LCN2 in inflammatory injury and tumorigenesis, the relationship between LCN2 and ferroptosis in sepsis-induced liver injury requires further investigation.
In summary, LCN2 signaling is activated in the model of sepsis-induced liver injury, thereby increase the cytoplasmic LIP and facilitating cell ferroptosis of hepatocyte, which eventually exacerbates liver injury. Inhibition of LCN2 expression is thus expected to provide a new strategy for the clinical treatment of liver injury in sepsis and a theoretical basis for the prevention and treatment of sepsis.
5ConclusionsDuring liver injury in septic mice, inhibition of ferroptosis and iron overload may alleviate liver injury, and reduced LCN2 expression may protect the liver against sepsis-induced damage by reducing ferroptosis mediated by xCT and DHODH. Ferroptosis may be the key to improving sepsis-induced liver injury, and LCN2 represents a potentially effective target for its treatment. However, the underlying mechanism needs to be investigated further.
FundingThis work was supported by Natural Science Research Project of Anhui Province Education Department [2022AH040213], and Graduate Research Innovation Program of Bengbu Medical University [Byycx22001].
Author contributionsYuping Li conceived the study, fulfilled data acquisition, analysis and write the manuscript, Lu Li conducted the statistical analysis, Yuming Zhang, Qi Yun and Ruoli Du participated in the animal experiments, Hongwei Ye revised the manuscript, Zhenghong Li participated in the experimental design, Qin Gao designed the experiment protocol and provided the support for the project. All authors have read and agreed to the published version of the manuscript.
Data availabilityThe datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.



