



Hepatocellular carcinoma is among the most frequent forms of primary liver cancer and develops within a context of chronic inflammation, frequently associated with a multitude of risk factors, including viral infections, metabolic dysfunction-associated fatty liver disease, metabolic dysfunction-associated steatohepatitis and liver fibrosis. The tumor microenvironment is crucial for the progression of HCC, as immune cells, tumor-associated fibroblasts and hepatic stellate cells interact to promote chronic inflammation and tumor spread. Inflammasomes, the multiprotein complexes that launch the innate immune response, emerge as important mediators in the pathogenesis of HCC. Among others, the inflammasome Nucleotide-binding oligomerization domain, Leucine rich Repeat (NLR) and Pyrin (NLRP) 3 (NLRP3), and absent in melanoma 2 (AIM2), exhibit a dual role in HCC background. It has been reported that they can exert oncosuppressive functions by triggering the inflammatory death of cancer cells. Vice versa, chronic activation contributes to the development of a pro-tumorigenic environment, thus supporting tumor growth. In addition, other inflammasomes such as Nucleotide-binding oligomerization domain, Leucine rich Repeat (NLR) and Pyrin (NLRP) 6 and 12 (NLRP6 and NLRP12, respectively) regulate HCC onset and progression, although more experimental evidence is required. This review focuses on the molecular mechanisms underpinning the inflammasome's contribution to the onset, progression and spread of HCC. Moreover, we will explore the potential therapeutic approaches currently under investigation, which aim to improve the efficacy and reduce the side effects of the treatments currently available. Targeting inflammasomes may be a promising therapeutic strategy for the treatment of HCC, offering new opportunities to improve patient prognosis.
Hepatocellular carcinoma (HCC), which has an incidence of 90% of cases, remains the major form of primary liver cancer [1], being estimated as the fifth and sixth most frequent tumor in males and females, respectively [2,3]. Primary risk factors that contribute to HCC development, such as hepatitis B (HBV) and hepatitis C (HCV) viruses, cirrhosis, alcoholism, exposure to aflatoxins, genetic and epigenetic alterations, microRNA (miRNA) disorders, altered programmed cell death pathways, the epithelial-to-mesenchymal transition (EMT), increased HCC stem cells, are nowadays well-known, alongside the chronic inflammatory niches characteristic of this neoplasm [4,5]. The pathogenesis of HCC falls, historically, into two main recognized sub-groups, proliferative, with a poorer clinical outcome [6,7], and non-proliferative. Malignant cell proliferation can involve the constitutive activation of Wingless-related integration site (WNT)-β-Catenin, Phosphoinositide 3-Kinase (PI3K)-Protein Kinase B (AKT)-mechanistic Target Of Rapamycin (mTOR) and Rat sarcoma virus (RAS)-Mitogen-activated protein kinase (MAPK) pathways [8,9,10], owing to Fibroblast Growth Factor 19 (FGF19) amplification, as well as Axin1, Ribosomal protein S6 Kinase A3 (RPS6KA3) and/or Ribosomal S6 Kinase 2 (RSK2) mutations [11,12]. These arise upon the onset of somatic mutations, such as those that drive tumor protein p53 (P53) inactivation [11]; or follow Cyclin D1 gene (CCND1) amplifications [10] and α-Fetoprotein (AFP) overexpression [13], whereas β-Catenin gene (CTNNB1) and Telomerase Reverse Transcriptase (TERT) promoter mutations belong to the nonproliferation class [14,15]. Moreover, in the last decade, a strong relationship has been identified between HCC development and an altered glucose and lipid metabolism, which, in turn, determines metabolic disorders such as obesity, type two diabetes mellitus and hepatic steatosis. Acting as a bridge to chronic inflammation, this condition leads to a clinical picture contextualized as metabolic dysfunction-associated fatty liver disease (MAFLD) [16], which affects roughly 40% of the world population and is the major contributor to HCC development [17]. Another ingredient that determines hepatocarcinogenesis is the tumor microenvironment (TME), assembled by a variety of cell types, encompassing hepatic stellate cells (HSCs), which sustain tumor spread via inflammatory cytokines and growth factors release [18], and promote chemoresistance [19]; endothelial cells (ECs) and cancer-associated fibroblasts (CAFs), involved in the angiogenesis process through the secretion of Vascular Endothelial Growth Factor (VEGF), Matrix Metalloproteinase 2 (MMP2), Matrix Metalloproteinase 9 (MMP9), Epidermal Growth Factor Receptor (EGFR), Platelet-Derived Growth Factor Receptor (PDGFR), C-X-C Chemokine Receptor (CXCR) and Transforming Growth Factor β (TGF-β) factors [20]; innate immune cells, such as M1 (classic) or M2 (alternative) macrophages and tumor-associated macrophages (TAMs), which, although they act through different, and sometimes still controversial interventions, are both able to foster HCC development and progression. In particular, while M2 macrophages are programmed to release growth factors and provide an immunosuppressive phenotype, M1 generate a pro-inflammatory environment and encourages oncogenic mutations [21] in an inflammasome-dependent fashion [22]. Only relatively recently identified, the inflammasome is an intracellular multiprotein complex that triggers Caspase-1 activation, which, in turn, allows the maturation and release of pro-inflammatory cytokines, such as interleukin-1β (IL-1β) and interleukin-18 (IL-18), upon detection of harmful stimuli, thus playing a key role in the body's immune response [23]. However, excessive inflammasome activation is often a mechanism employed by M1 macrophages within the tumor microenvironment to nourish all the stages of tumorigenesis [24]. Although some studies report a cancer-suppressive role for the inflammasome [25], the majority describe a more pronounced neoplastic scenario [26]. Unresolved inflammatory foci within the TME, therefore, allow malignant cells to elude the immune surveillance, a situation that is discernible by means of the abundance of regulatory T (Tregs) and myeloid-derived suppressor cells (MDSCs) [27]. For this reason, the inflammasome and its effector cytokines, have been designated a promising target for HCC therapy. In this review, we summarize the current knowledge of different characterized inflammasomes and their contribution to the onset, progression, and metastasis of HCC. We will also explore potential revolutionary pharmaceutical approaches that could enhance the currently approved treatments for HCC.
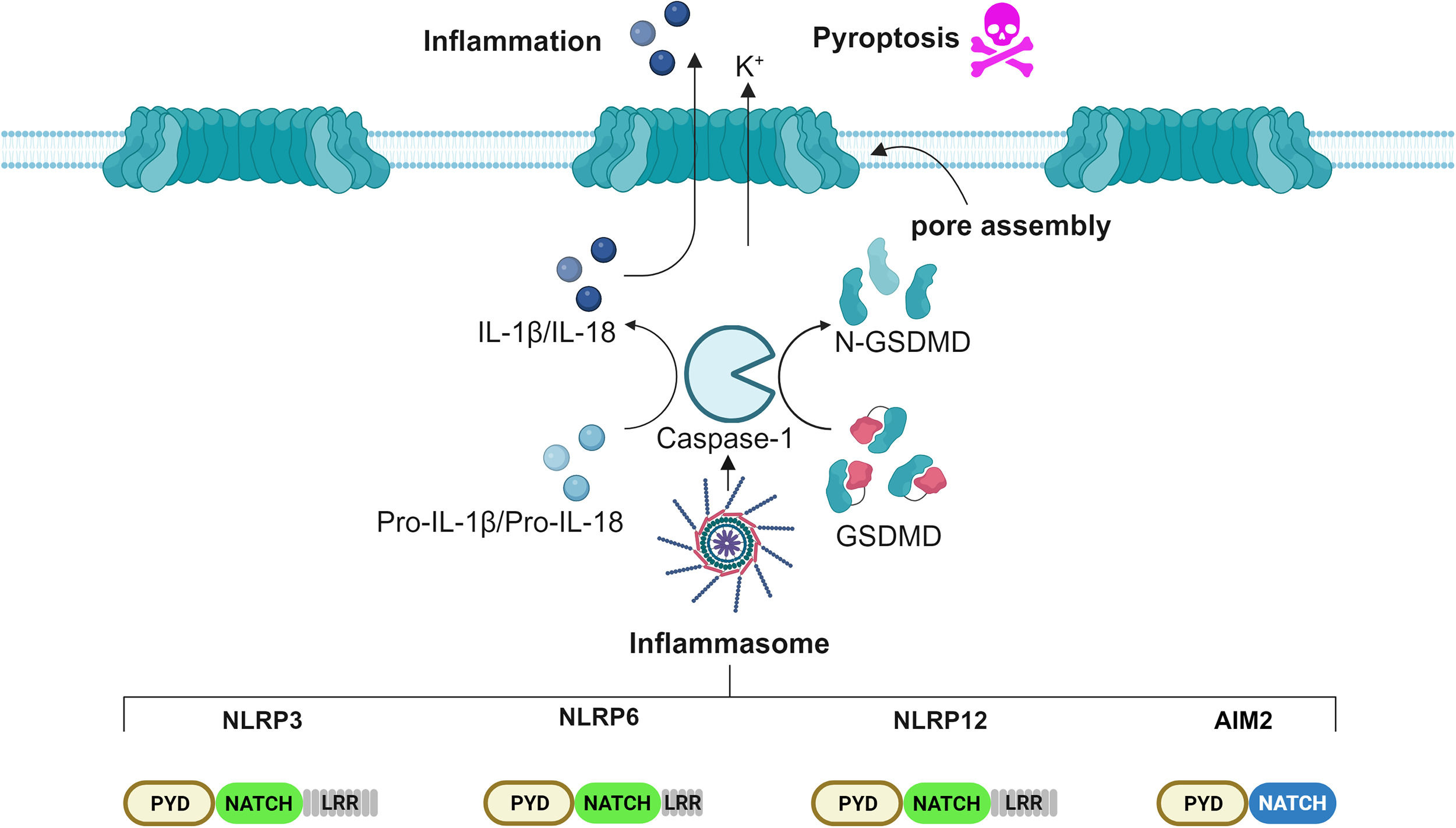
2Inflammasome: Novel insights into the molecular mechanism of activationPattern recognition receptors (PRRs) are an essential component of the innate immune response to injury. PRRs-recognized threats are classified in two main categories: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) [28]. Following detection, PRRs assemble and signal to an intracellular complex denominated the inflammasome, which organizes the inflammatory response [29]. Structurally, inflammasomes comprise three groups: the Nucleotide-binding oligomerization domain, Leucine rich Repeat (NLR) and Pyrin (NLRP) or Card (NLRC) domain-containing inflammasomes, absent in melanoma 2 (AIM2)-like receptor (ALR)-associated inflammasome, and the Pyrin inflammasome. The broad spectrum of inflammasomes allows the detection of a vast range of microbial inputs by both immune cells, such as monocytes, macrophages and dendritic cells, as well as by non-immune cells, such as intestinal epithelial cells and fibroblasts [30,31]. The human NLR family encompasses 22 members, among which the best characterized is the NLRP3 inflammasome [32]. Upon recognition of a threat, PRRs bind to the apoptosis-associated speck-like protein containing a CARD (ASC), which, in turn, recruits and activates pro-Caspase-1, assembling the core of the inflammasome. The cascade proceeds with the activation, mediated by Caspase-1, of pro-IL-1β, pro-IL-18 and Gasdermin D (GSDMD) into their bioactive form. Cleaved GSDMD N-terminus domains auto-assemble into wheel-shaped structures, migrate onto the plasma membrane and, together with nerve injury-induced protein 1, named ninjurin-1 (NINJ1), form pores, allowing the release of mature cytokines IL-1β and IL-18 into the bloodstream, as well as a form of inflammatory cell death called pyroptosis (Figure 1). Microscopically detectable as a single punctum or "speck" per cell, the inflammasome has a diameter ranging from 1 to 3 μm [33]. Although considered beneficial, inflammasome activation must be tightly regulated. Undesired activation could, in fact, contribute to the worsening of several inflammation-driven diseases, such as cryopyrin-associated periodic syndrome, arthritis, atherosclerosis, type 2 diabetes, Alzheimer's disease, inflammatory bowel diseases (IBDs), including Crohn's disease (CD), ulcerative colitis (UC), and cancer [34]. The NLRP3 inflammasome is the best characterized nowadays, displaying both protective and pathogenic traits related to the gastrointestinal tract [35]. The current knowledge of the NLRP3 inflammasome includes three different models of activation, tagged as canonical, non-canonical and alternative activation [36]. The canonical model involves two temporally distinct steps, denominated priming and activation. During the priming step, microbial insults, such as lipopolysaccharides (LPS), are sensed by the Toll-like receptors (TLRs), which transduce the signal to a large intracellular oligomeric signaling complex, termed Myddosome. The Myddosome complex consists of the adaptor (MYD88-adapter-like) MAL, Myeloid Differentiation Primary Response 88 (MYD88), Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) and Interleukin-1 Receptor-Associated Kinase 1/2 (IRAK1/2) and is appointed to induce Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-kB) activation, which, in turn, enhances the cytosolic level of the pro-inflammatory cytokines IL-1β and IL-18 [37]. The priming step is then followed by the activation step. This occurs when a second threat is perceived by the NLRP3 inflammasome and culminates in full inflammasome activation. Numerous chemical and physical stimuli have been ascribed an involvement in the activation step, such as nigericin, muramyl dipeptide, adenosine triphosphate (ATP), metabolic stress, alterations in cell volume, uric acid crystals, silica, asbestos, calcium phosphate, alum, potassium efflux, lysosomal destabilization, increased levels of reactive oxygen species (ROS), oxidized mtDNA (ox-mtDNA), trans-Golgi rupture [38]. In addition, post-translational modifications (PTMs), such as phosphorylation, ubiquitination and sumoylation, are required for NLRP3 activation [39]. Once activated, it migrates to the microtubule-organizing center (MTOC) at the perinuclear region, where it encounters and binds to NIMA Related Kinase 7 (NEK7), to promote ASC nucleation [33]. Non-canonical NLRP3 activation bypasses the priming step and involves a direct binding between cytosolic LPS and Caspase-4/5. Active Caspase-4/5 induces a potassium efflux, which triggers the NLRP3 inflammasome activation [40]. Lastly, alternative NLRP3 activation engages Caspase-8 upon LPS internalization [41]. Recent studies have demonstrated that the NLRP3 inflammasome plays a crucial role in the pathogenesis of various liver diseases by promoting chronic inflammation, fibrosis, cirrhosis, and tumorigenesis [42]. Besides the bona fide inflammasome NLRP3, immune and non-immune cells harbor the AIM2 inflammasome, which detects self- and non-self-cytoplasmic double-stranded DNA (dsDNA), released to counteract cellular stress or pathogenic infection [43]. Structural studies of AIM2 revealed that the protein architectural domain guarantees a certain degree of auto-inhibition in quiescent conditions that are disrupted upon binding to dsDNA. Moreover, post-translational modifications, such as ubiquitination, ensure inappropriate activation. Following dsDNA detection, Ubiquitin-Specific Protease 21 (USP21) deubiquitinases AIM2, thus allowing its fast nucleation [44]. So far, several inflammasomes have been linked to the development of HCC: NLRP3, AIM2, NLRP6 and NLRP12, while NLRP3 and AIM2 inflammasomes perform tumor-suppressing as well as tumor-promoting functions [45].

Scheme of the inflammasome pathway involved in HCC. Several studies show that the inflammasomes NLRP3, NLRP6, NLRP12 and AIM2 play roles in HCC. These inflammasomes act by cleaving Caspase-1, which in turn activates the pro-inflammatory cytokines, IL-1β and IL-18, alongside the pore-forming protein GSDMD, which ultimately promotes pyroptosis. Prolonged inflammasome activation leads to tissue inflammation.
Due to the lack of effective therapeutic treatments, HCC exhibits poor prognosis, particularly in patients with advanced-stage cancer [46,47]. Investigating the mechanisms underlying HCC development is essential to identify novel and effective therapeutic approaches. A large volume of scientific evidence nowadays imputes a central role of the NLRP3 inflammasome in HCC by affecting various aspects of cancer onset, such as growth, invasiveness, angiogenesis and spread [48,49], whereas other works have reported anti-neoplastic effects. Several factors, including tumor stage, cellular context and the balance between pro-inflammatory and anti-inflammatory cytokines, may influence the role of the NLRP3 inflammasome in HCC progression [50] (Figure 2). Nonetheless, further studies are needed to understand the mechanisms underlying the transition from oncosuppressor to oncopromoter functions of the NLRP3 inflammasome in a context-specific manner.
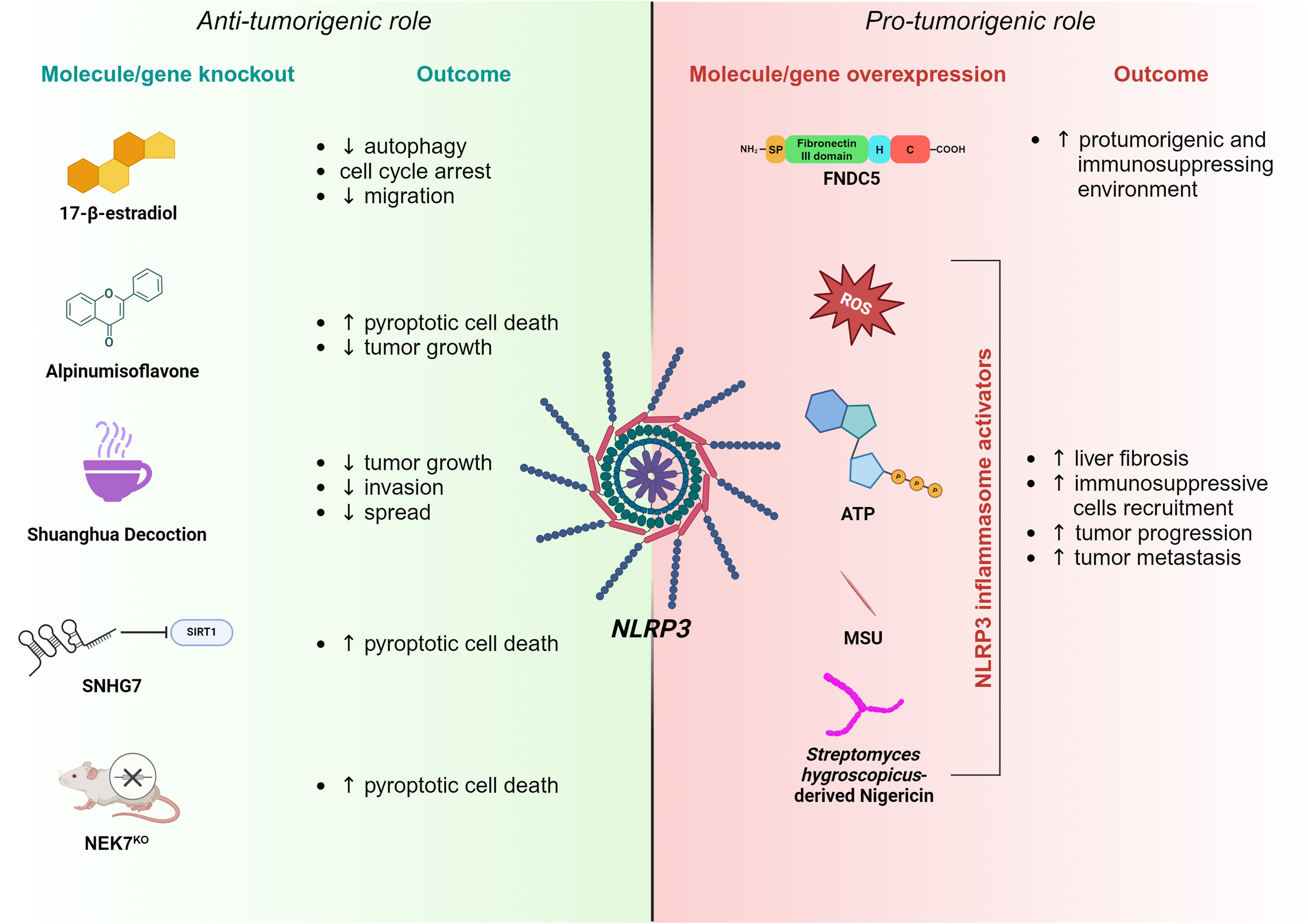
 and a pro-tumorigenic role (right panel) in HCC progression. Anti-tumorigenic features of NLRP3 are exerted by 17-β-estradiol, Alpinumisoflavone, Shuanghua Decoction, SNHG7 or NEK7 knockout, having different negative effects on tumor growth and progression. On the contrary, the overexpression of FNDC5 or NLRP3 inflammasome activators induces pro-tumorigenic traits through the NLRP3 pathway.")
Schematic of NLRP3 inflammasome involvement in HCC. Accumulating evidence suggests that the NLRP3 inflammasome may have both an anti-tumorigenic (left panel) and a pro-tumorigenic role (right panel) in HCC progression. Anti-tumorigenic features of NLRP3 are exerted by 17-β-estradiol, Alpinumisoflavone, Shuanghua Decoction, SNHG7 or NEK7 knockout, having different negative effects on tumor growth and progression. On the contrary, the overexpression of FNDC5 or NLRP3 inflammasome activators induces pro-tumorigenic traits through the NLRP3 pathway.
Among other functions, autophagy has been recognized to exert pivotal roles in HCC onset and development. In particular, it shows proliferative features while inhibiting apoptosis. Moreover, the induction of autophagy promotes tumor metastasis via the EMT and regulates HCC stem cell formation [51]. Interestingly, NLRP3 inflammasome activation downregulates autophagy via the 17β-estradiol/Estrogen Receptor Beta/Adenosine Monophosphate-Activated Protein Kinase/ mechanistic Target Of Rapamycin (E2)/ERβ/AMPK/mTOR) pathway, thereby suppressing cancer development [52]. Following inflammasome activation on Bel-7402 and SMMC-772 HCC cell lines, Dai and colleagues observed an arrest of the cell cycle in the Gap1 (G1) phase alongside the inhibition of their migratory attitude [53]. Among natural compounds, alpinumisoflavone (AIF) has been studied for its cytotoxic and anti-metastatic properties in several cancers because of its ability to block cell cycle progression and promote apoptosis. In a study conducted by Zhang et al., the treatment of various HCC cell lines with AIF triggered NLRP3-mediated pyroptosis, affecting tumor growth. This phenotype has been further validated when autophagy was inhibited [54]. Traditional Chinese medicine offers a pharmacological recipe whose components exert anticancer effects by inhibiting tumor growth, inducing apoptosis, and stimulating the immune system [55,56]. This formula, called Shuanghua decoction (SHD), is mainly composed of Oldenlandia and Prunella spikes. SHD inhibited tumor growth, invasion, and spread in in-vitro experiments on HCC cell lines by inducing ROS production, NLRP3 inflammasome activation and preventing NF-kB nuclear translocation [57]. The long non-coding RNA (lncRNA) small nucleolar RNA host gene 7 (SNHG7) is overexpressed in liver cancer tissues and cell lines. It has been shown that SNHG7 silencing decreases Sirtuin 1 (SIRT1) levels, which, in turn, enhances NLRP3 inflammasome activation and, consequently, pyroptotic HCC cell death [58]. Surprisingly, genetic abrogation of NEK7, found to be upregulated in HCC tissue, promotes the expression of NLRP3, Caspase-1, and GSDMD, triggering pyroptosis of neoplastic cells [59].
3.2Pro-tumorigenic role of NLRP3Perpetuated inflammasome activation sustains the pro-tumorigenic niche by activating HSCs through the maturation and release of the effector cytokines, IL-1β and IL-18, which ultimately result in liver fibrosis [60]. Upon activation, NLRP3 inflammasome triggers ROS and cathepsin B activity in HSCs, which, in turn, induces the expression of several markers of fibrosis. These findings were further supported by NLRP3-deficient mice experiments, where the authors demonstrated a less pronounced fibrotic phenotype [61]. Many studies have shown that NLRP3 activation, followed by IL-1β secretion, recruits immunosuppressive cells into the TME, including MDSCs and TAMs to boost tumor progression and metastasis onset [62]. Overexpression of the Fibronectin Type III structural domain-containing protein 5 (FNDC5), a cytokine involved in processes regulating energy metabolism, alters the Peroxisome Proliferator-Activated Receptor Gamma/ Nuclear Factor kappa-light-chain-enhancer of activated B cells/ Nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin 3 (PPARγ)/NF-kB/NLRP3 pathway, triggering the M1 (classically activated macrophage)/M2 (alternatively activated macrophage) phenotype switch, which fosters an immunosuppressive and pro-tumorigenic environment [63,64]. Moreover, NLRP3 inflammasome activation affects the anti-tumor immune surveillance in HCC, commonly exerted by natural killer (NK) cells. The immune response is known to operate through the binding between the Natural killer Group D (NKG2D) receptors, expressed on the NK cell surface, and their ligand, the Major Histocompatibility Class (MHC) I chain-related protein A/B (MICA/B), found on HCC cells [65,66,67,68]. Extensive screening of human HCC tissue correlated the expression of NKG2D receptors with improved patient survival [69], although the molecular mechanism involved still needs to be elucidated. In addition, Lee and colleagues have recently shown that NLRP3 knockout HCC cells display an increased expression of MICA/B on their surface, resulting in enhanced NK cell cytotoxicity and reduced tumor growth and metastasis [70]. Consistently, TAMs co-cultured with HCC cells engaged the IRAK1-NLRP3 signaling axis, fostering their proliferative and migratory abilities through a positive feedback loop involving IL-1β, IL-18 and monocyte chemoattractant protein 1 (MCP-1). On the contrary, IRAK1 deficient HCC cells abolished TAMs influence on tumor development [49]. Other independent groups have described several molecules affecting the activity of the NLRP3 inflammasome on HCC. By binding the 3′-untranslated region (3’-UTR) of NLRP3, the tumor suppressor miR-223-3p inhibits its activation, ultimately promoting neoplastic cell apoptosis while inhibiting proliferation [71]. Similarly, luteoloside, a flavonoid isolated from Gentiana macrophylla, was capable of reducing ROS accumulation and thus inhibiting tumor progression via the NLRP3 inflammasome [72]. Overall, these data list NLRP3 as a potential target for a new therapeutic strategy in HCC treatment.
4Role of the AIM2 inflammasome in HCC developmentResearch conducted over the last decade has revealed that AIM2 plays essential roles in inflammatory-driven diseases such as HCC, MAFLD, metabolic dysfunction-associated steatohepatitis (MASH), hepatitis B and liver fibrosis through inflammasome-related and/or unrelated mechanisms. [73,74]. Like NLRP3, the AIM2 inflammasome displays both anti- and pro-tumorigenic functions against the HCC background (Figure 3).
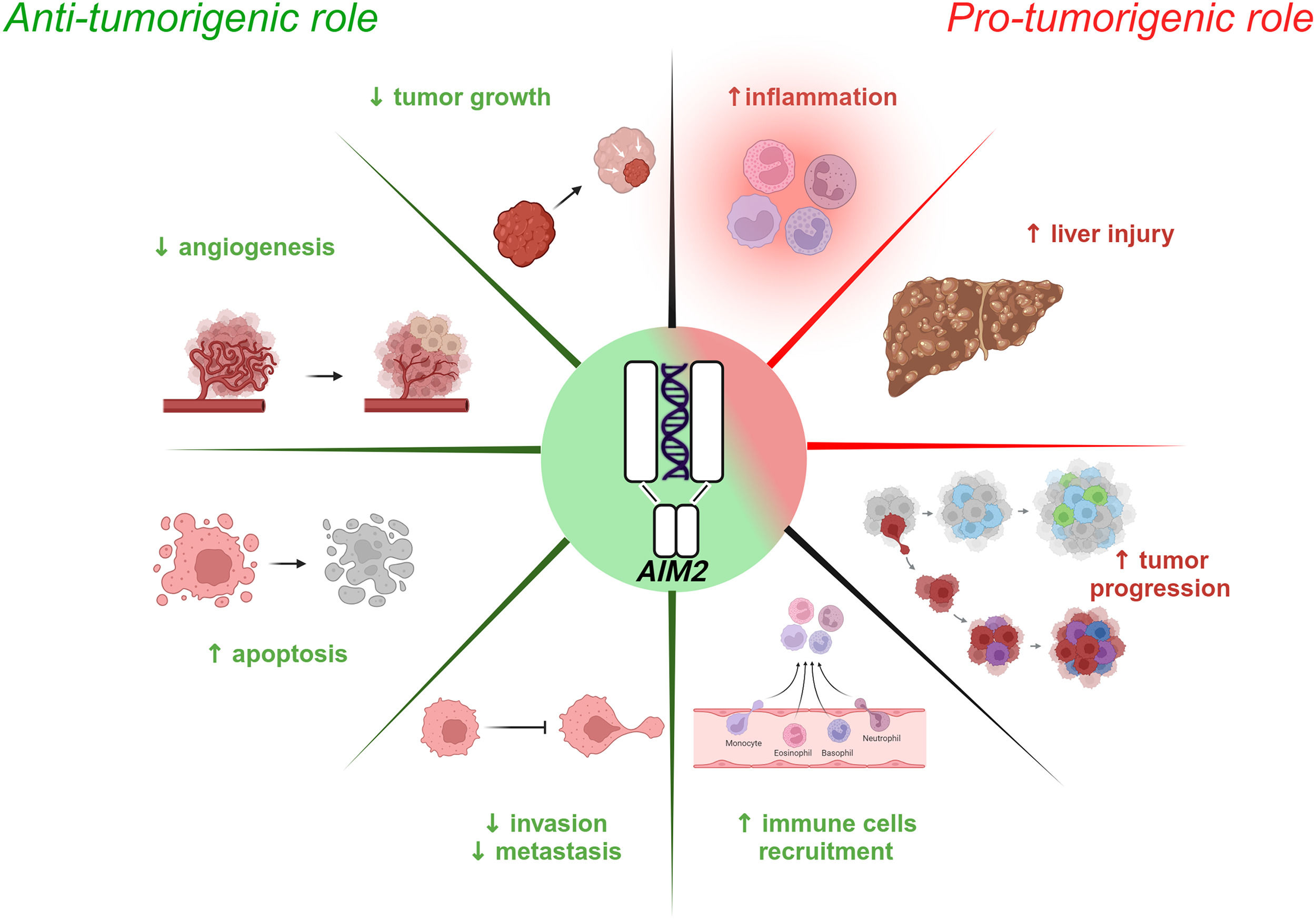
 and pro-tumorigenic (right panel) effects. Activated AIM2 exerts anti-tumorigenic effects by suppressing tumor growth, angiogenesis, invasion and metastasis while promoting the recruitment of immune cells and inducing apoptosis of HCC cells. Conversely, AIM2 pro-tumorigenic activity is characterized by exacerbated inflammation, liver damage and tumor progression.")
Schematic of AIM2 inflammasome involvement in HCC. Emerging evidence highlights the dual role of the AIM2 inflammasome in HCC, exhibiting both anti-tumorigenic (left panel) and pro-tumorigenic (right panel) effects. Activated AIM2 exerts anti-tumorigenic effects by suppressing tumor growth, angiogenesis, invasion and metastasis while promoting the recruitment of immune cells and inducing apoptosis of HCC cells. Conversely, AIM2 pro-tumorigenic activity is characterized by exacerbated inflammation, liver damage and tumor progression.
Several studies have shown that AIM2 expression is significantly reduced in HCC tissue compared to healthy counterparts and that its downregulation is correlated with advanced cancer progression [75]. Some evidence, in fact, supports a tumor-suppressive role for AIM2 by restraining the mTOR-S6K1 pathway in HCC cells and, consequently, limiting the expression of the Hypoxia-Inducible Factor (HIF-1α), involved in tumor growth, angiogenesis, and cancer cell metabolism [76,77]. A lack of AIM2 reflects mTOR-S6K1 pathway overactivation, promoting HCC development; this is correlated with larger tumors and increased tumor spread [Ma et al., 2016]. On the contrary, AIM2 overexpression silences the mTOR-S6K1 pathway, thereby reducing cell proliferation, colony formation, and tumor cell invasiveness [75]. The HBV protein Hepatitis B Virus X (HBx) binds to the AIM2 promoter and induces its subsequent degradation. This leads to the enhancement of fibronectin 1 (FN1) with EMT activation and tumor spread [78]. Consistently, transcriptomic analyses and functional biochemistry and molecular biology assays performed by Zheng and colleagues indicated that reduced AIM2 levels are inversely associated with the expression of several EMT markers [79]. In addition, another group showed that AIM2-overexpressing HCC cells have a lower expression of genes related to the Notch pathway, which is known to negatively influence the tumor microenvironment [79]. Moreover, AIM2 inflammasome activation culminates with the release of pro-inflammatory cytokines, which in turn recruit immune cells such as B cells, Cluster of Differentiation 4 and 8 (CD4+ and CD8+, respectively) T cells, macrophages, neutrophils, and dendritic cells within the neoplastic tissue to nurture the immune response against the tumor [79].
4.2Pro-tumorigenic role of AIM2A few studies have reported a negative correlation between AIM2 deficiency and tumor progression, particularly in advanced stages of HCC. Prolonged inflammation leads to injury and continuous liver cell turnover, creating a favorable environment for tumor initiation [80,81,82]. Martínez-Cardona and colleagues found, in 2018, that diethylnitrosamine (DEN)-induced HCC mice displayed high levels of dsDNA in the serum, which triggered AIM2 activation and IL-1β-mediated tumor progression [83]. Although still controversial, a role has been suggested in the literature for AIM2 in HCC that seems to mirror the tumor stage, exerting protective features when the tumor is advanced and pro-tumorigenic functions in the early phase. Thus, additional studies are required to understand the relationship between HCC and AIM2 function, which might potentially include this inflammasome in the list of prognostic and/or predictive biomarkers of this pathological scenario.
5Roles of other inflammasomes in HCC onset and progressionAlongside NLRP3 and AIM2, other inflammasomes may be involved in the pathogenesis of HCC. Recent studies have demonstrated that NLRP6 overexpression is correlated with better patient survival, lower levels of detrimental prognostic markers, such as AFP and enhanced immune cell infiltration within the TME [84]. Conversely, in vivo, experiments have shown that abnormal intestinal Candida albicans (C. albicans) colonization might enhance HCC through the involvement of the NLRP6 inflammasome [85]. Therefore, more studies are required to elucidate the exact role of NLRP6 against the HCC background. NLRP12 plays several protective roles in HCC by interfering with the c-Jun N-terminal kinase (JNK) pathway, which is known to foster cancer cell proliferation. NLRP12-deficient mice display unrestrained JNK activation, with high levels of cellular Jun (c-Jun) and cellular Myelocytomatosis (c-Myc) oncogenes, which are correlated with HCC aggressiveness. The authors concluded that NLRP12 exerts a negative regulation of the JNK pathway, preventing inflammation and HCC cell proliferation [86,87].
6Therapeutic approaches targeting the NLRP3 and AIM2 inflammasomes in HCC treatmentCurrently approved therapies for hepatocellular carcinoma (HCC) consist of several choices, ranging from surgical resection to liver transplantation, local ablation techniques, site-specific treatments, and systemic chemotherapy [88,89]. However, for patients with advanced HCC, the efficacy of these treatments is poor and the prognosis remains unfavorable. Sorafenib, a multikinase inhibitor, is one of the main systemic drugs used as targeted therapy for this type of malignancy [90,91]. In recent years, however, advances in research have significantly improved therapeutic options for HCC, with significant effects on clinical outcomes (Table 1). Among the most promising innovations, immunotherapy and nanocarriers have emerged as two approaches that can profoundly change the treatment of this type of cancer. Such methods have the potential not only to improve the response to treatment but also to reduce systemic toxicity, a common side effect of traditional therapies [92;93], thus opening the door to more personalized and less invasive treatment protocols [94,95]. As reported above, the roles of the NLRP3 and AIM2 inflammasomes in HCC are conflicting and demonstrate pivotal roles promoting either anti- or pro-tumorigenic phenotypes. Several compounds have been tested through in vitro and in vivo strategies targeting the inflammasome in the context of HCC. For instance, miR-223-3p and luteoloside, by lowering NLRP3 expression, inhibit cell proliferation, reduce the migratory capacity and suppress HCC growth [71,72]. Meanwhile, E2, AIF and SHD, induce NLRP3 activation, promote pyroptosis and suppress HCC proliferation, growth, invasion and metastasis. [52,53,54]. Besides these molecules, other studies have investigated NLRP3-targeted therapies for HCC. A recent study found that mice treated with a specific Peroxisome Proliferator-Activated Receptor α (PPARα) agonist, pirinixic acid (WY-14643), upregulated the expression of Gm15441 lncRNA, known to inhibit thioredoxin-interacting protein (TXNIP) levels, and thus prevent NLRP3 inflammasome activation. WY-14643 reduced inflammation and oxidative stress in the liver, showing it to be a suitable target for the treatment of HCC [96]. HCC-bearing mice treated with a combination of metformin and celastron, an NLRP3 inhibitor, had reduced levels of IL-1β and IL-18, alongside lower neoplastic cell proliferation and angiogenesis, while liver functions were improved [97]. Furthermore, in syngeneic and subcutaneous mouse models of HCC, inhibition of the NLRP3 inflammasome enhanced the antitumor activity of sorafenib. Specifically, the combination of sorafenib and MCC950, another selective and potent NLRP3 antagonist, increased the tumor necrosis area and downregulated Proliferating Cell Nuclear Antigen (PCNA) expression (a marker of cell proliferation) without any apparent liver injury [98]. Like NLRP3, AIM2 may be a potential target for HCC therapy. Experimental evidence suggests that the antimalarial drug dihydroartemisinin (DHA), induces autophagy and HCC cell death by upregulating the AIM2 inflammasome and promoting pyroptosis of neoplastic cells [99]. Moreover, radiofrequency ablation treatment significantly increased the levels of AIM2, Caspase-1 and the pro-inflammatory cytokines IL-1β and IL-18 in nude HCC xenograft mice compared with control groups. Although the immunological mechanism still needs to be clarified, this led to AIM2-mediated pyroptosis of HCC cells [100]. Finally, the identification of new therapeutic targets in cancer treatment may have a critical role in leading to precision oncology, improving patient outcomes, and overcoming barriers to conventional therapies.
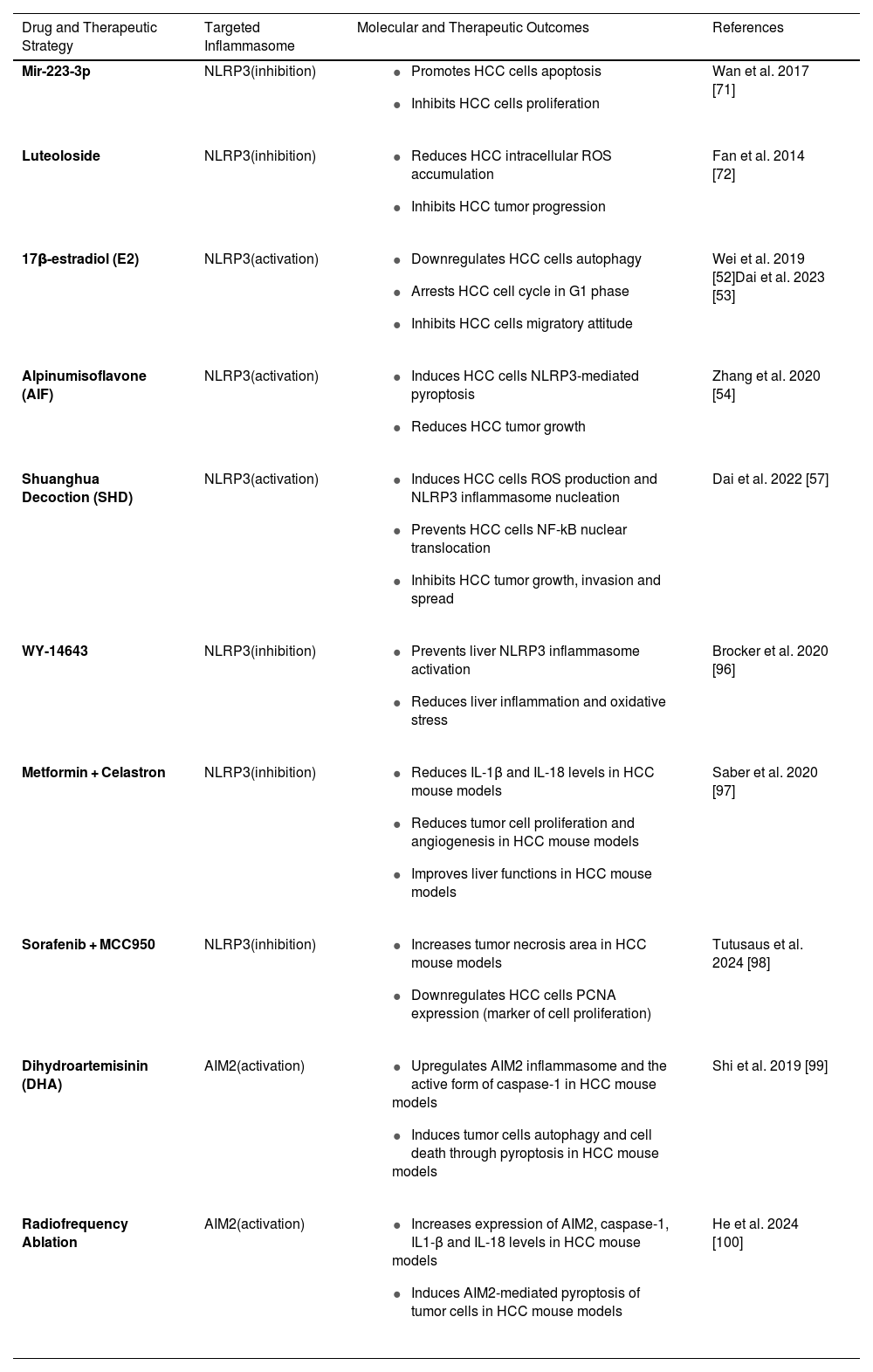
Summary of the molecular targets and therapeutic outcomes in HCC.
| Drug and Therapeutic Strategy | Targeted Inflammasome | Molecular and Therapeutic Outcomes | References |
|---|---|---|---|
| Mir-223-3p | NLRP3(inhibition) |
| Wan et al. 2017 [71] |
| Luteoloside | NLRP3(inhibition) |
| Fan et al. 2014 [72] |
| 17β-estradiol (E2) | NLRP3(activation) |
| Wei et al. 2019 [52]Dai et al. 2023 [53] |
| Alpinumisoflavone (AIF) | NLRP3(activation) |
| Zhang et al. 2020 [54] |
| Shuanghua Decoction (SHD) | NLRP3(activation) |
| Dai et al. 2022 [57] |
| WY-14643 | NLRP3(inhibition) |
| Brocker et al. 2020 [96] |
| Metformin + Celastron | NLRP3(inhibition) |
| Saber et al. 2020 [97] |
| Sorafenib + MCC950 | NLRP3(inhibition) |
| Tutusaus et al. 2024 [98] |
| Dihydroartemisinin (DHA) | AIM2(activation) |
| Shi et al. 2019 [99] |
| Radiofrequency Ablation | AIM2(activation) |
| He et al. 2024 [100] |
AIF, Alpinumisoflavone; AIM2, Absent in melanoma 2; Caspase-1, A protease enzyme playing a role in inflammasome activation; DHA, Dihydroartemisinin; E2, 17β-estradiol (a form of estrogen); HCC, Hepatocellular carcinoma; IL-1β, Interleukin-1 beta; IL-18, Interleukin-18; miR-223-3p, MicroRNA-223-3p; NF-κB, Nuclear factor kappa B; NLRP3, NOD-like receptor family pyrin domain-containing 3; PCNA, Proliferating cell nuclear antigen; ROS, Reactive oxygen species; SHD, Shuanghua Decoction; WY-14643, A peroxisome proliferator-activated receptor-alpha (PPAR-α) agonist.
The relatively new branch of innate immunity, identified as inflammasomes, has gained substantial attention nowadays, especially in cross-field research such as immune-oncology. The present review aims to summarize the latest understanding of the role of the inflammasome in the onset and development of HCC. Inflammasomes are involved in a large spectrum of common diseases and display tissue-contextualized functions. However, the consequences of inflammasome activation and GSDMD-mediated pyroptosis are still contradictory in the oncogenic field, as some studies have reported pro-tumorigenic episodes and others anti-tumorigenic effects. Elevated levels of bioactive pro-inflammatory cytokines, IL-1β and IL-18, shape the local environment, inducing distinct outcomes, including the onset and progression of HCC. Hence, a balanced inflammasome-pyroptosis axis activation is essential to preserve proper homeostasis. Accordingly, depending on the degree of inflammasome activation, the response to therapy is also partially affected [101]. Although research into the role of the inflammasome in cancer is still in its early stages, advances in research technologies, nanomaterials, and both natural and chemotherapeutic drugs offer significant promise in this area. Exciting approaches focus on targeting two key components of the tumor microenvironment: TAMs and CAFs, which sustain HCC progression through the activation of the inflammasome [102]. Moreover, novel perspectives blocking cytokine release, inactivating inflammasome through post-translational modifications and shifting the proliferative propensity of cancer cells towards a pro-apoptotic phenotype should be considered, offering a therapeutic advantage for selectively targeting malignant cells. Importantly, this sub-field of the immune-oncology area has started to capture the interest of numerous biotech companies, which are eagerly conducting high-throughput screening to identify novel inflammasome activators. However, to a certain extent, due to the genotypic and phenotypic heterogeneity of various tumors, cancer cells are vulnerable to inflammasome-mediated cell death. Mutations or changes in the expression levels of inflammasome components may impact treatment efficacy in HCC subjects. In summary, a deeper knowledge would also be beneficial for advancing personalized medicine clinical applications, potentially integrating the inflammasomes as prognostic biomarkers and therapeutic targets in HCC treatment.
FundingThis research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Authors contributionVA conceived the concept and wrote the initial draft of the manuscript. RN and GG edited the manuscript and arranged the layout. VA and RN drew the figures.
Figures 1, 2 and 3 in this manuscript were created with BioRender software (https://app.biorender.com/).



