



Tumor-associated macrophages (TAM) can influence both cancer growth and chemoresistance, but the specific mechanisms involved in these processes in cholangiocarcinoma (CCA) are unclear.
Materials and MethodsWe explored the distribution of TAM in CCA samples by multiplex immunofluorescence staining and tested the effects of TAM on CCA in vitro and in vivo. We then investigated the mechanisms underlying these effects using the Luminex assay, RNA sequencing, western blotting, flow cytometry, and co-immunoprecipitation.
ResultsThe infiltration of TAM was strongly increased in the cholangiocarcinoma tumor microenvironment. Oncostain M (OSM) secreted by TAM increased the proliferation and chemotherapeutic resistance of CCA cells both in vitro and in vivo. The results of transcriptome sequencing analysis, Western blot analysis, and immunofluorescence staining confirmed that OSM can promote Yap nuclear translocation and its subsequent formation of complexes with SMADs to upregulate the expression of inhibitor of DNA binding 1 (ID1).
ConclusionsTAM promotes CCA progression and chemoresistance through activating OSM-Yap-ID1.
In recent years, the incidence of cholangiocarcinoma has significantly increased worldwide, with cholangiocarcinoma currently accounting for 10 %-15 % of malignant liver tumors. The subtle early symptoms and rapid progression of CCA tumors lead to the loss of curative surgical opportunities for most patients [1,2]. However, even with the use of curative surgical methods, the 5-year overall survival rate is only 20 %-35 % [3]. The chemotherapy regimen containing gemcitabine and cisplatin has been the most effective first-line treatment for patients with unresectable cholangiocarcinoma in the last decade [4]. Moreover, many cholangiocarcinoma patients develop drug resistance shortly after chemotherapy, highlighting the urgent need to explore the mechanisms of chemoresistance to improve treatment efficacy [5].
The cholangiocarcinoma tumor microenvironment (TME) harbors various cells, including various immune cells, fibroblasts, and endothelial cells, among which tumor-associated macrophages (TAM) are a distinctive type. Macrophages exhibit plasticity and cells in the tumor microenvironment can participate in regulating macrophage education, leading to diverse phenotypes. Compared with adjacent normal tissues, most tumor-associated macrophages in the microenvironment of cholangiocarcinoma showed an alternative activation (M2) phenotype [6]. Many studies have confirmed that TAM can activate multiple pathways through secreting cytokines to promote the progression, metastasis, and treatment resistance of cholangiocarcinoma. The high density of TAM in the tumor microenvironment is associated with adverse clinical outcomes in various cancers [7,8]. Interestingly, many researchers suggest that TAM plays a crucial role in tumor progression and treatment resistance by regulating epithelial–mesenchymal transition (EMT) [9]. EMT is a process that endows tumor cells with mesenchymal stem cell-like characteristics and maintains tumor stemness, and it is primarily associated with tumor progression and drug resistance [10]. Studies have confirmed that TAM induces EMT in liver cancer cells by secreting TGF-β and increasing cellular invasive capabilities [11]. Additionally, M2 macrophages secrete TGF-β, which induces cancer cell EMT and chemoresistance through the aPKCɩ-NF-κB signaling pathway in CCA [12]. However, the role and mechanisms of crosstalk between TAM and cancer cells during EMT process in CCA remain incompletely understood.
DNA-binding inhibitor proteins (ID) are members of the helix-loop-helix family of proteins, which are involved in regulating the cell cycle and cell differentiation [13]. ID1, a stem cell-like gene, is overexpressed in various tumors and participates in several classical cancer-related signaling pathways, including EMT-related pathways, STAT3 signaling, and the TGF-β pathway [14]. Previous research has shown high expression of ID1 in non-small cell lung cancer, where it promotes tumor progression and metastasis by activating EMT [15]. Additionally, high ID1 expression plays a role in resistance to various treatments, including chemotherapy, radiotherapy, and targeted therapy. Studies have shown that elevated ID1 expression upregulates the tumor stemness markers Nanog and Oct4, accelerating the development of resistance to cisplatin in gastric cancer cells [16]. ID1 is a promising therapeutic target; crizotinib can inhibit cell migration by reducing ID1 levels in ALK- and MET-positive lung cancer cells, while γ-tocotrienol enhances chemotherapeutic efficacy in breast cancer cells by down-regulating ID1 expression [17,18]. However, the role and mechanisms of ID1 as a target in cholangiocarcinoma are not fully understood, and there is a lack of drugs targeting the ID1 pathway.
Numerous studies have confirmed the prominent role of the Hippo pathway in the initiation and progression of tumors. Although the main regulatory factors Yap and TAZ are rarely mutated in tumors, extensive research has revealed their ability to promote tumor progression, metastasis, and treatment resistance [19,20]. Yap is not considered an oncogene, but its high expression in tumor tissues has been confirmed to facilitate tumor cell proliferation and migration [21,22]. Additionally, the Hippo-Yap pathway has been found to typically interact and coordinate with other pathways in cancer research, such as the TGF-β signaling pathway, Wnt signaling pathway, and Notch signaling pathway. Studies have shown that the mutual interaction between the TGF-β/SMAD signaling pathway and the Hippo-Yap pathway significantly increases the malignancy of breast cancer cells [23]. However, no clear mechanism has been defined to clarify the roles of and interactions between these pathways in cholangiocarcinoma.
In summary, our study initially confirmed that TAM in the tumor microenvironment can secrete the cytokine OSM, promoting tumor cell proliferation and gemcitabine resistance. Furthermore, we discovered that OSM mediates the upregulation of ID1 through the key component YAP of the Hippo signaling pathway, thereby promoting EMT and increasing tumor stemness. These findings support the potential of investigating ID1 as a therapeutic target in cholangiocarcinoma.
2Materials and methods2.1Construction of cholangiocarcinoma organoidsTwo cholangiocarcinoma cell lines were obtained from the National Collection of Authenticated Cell Cultures. Cholangiocarcinoma cells were embedded in Matrigel at a ratio of 1:30, and the mixture was seeded in 48-well plates at 30 μl per well. After the mixture solidified, 500 μl of organoid culture medium was added to each well. The organoid culture medium was composed of advanced DMEM/F12, penicillin/streptomycin, Glutamax, B27 supplement, N2 supplement, HEPES, Gastrin, A83–01, Y-27,632, EGF, FGF10, R-Spondin1, Noggin and Afamin/Wnt3a CM.
2.2Tumor-associated macrophagesTAM were obtained using the Ficoll density gradient centrifugation method to isolate PBMCs. Then, PBMCs were initially cultured for 5 days in RPMI-1640 medium containing 10 % FBS and 20 ng/ml M-CSF. Subsequently, the culture was supplemented with a 40 % tumor cell-conditioned medium, and the cells were incubated for an additional 48 hours to obtain TAM.
2.3Construction of the organoids-TAM coculture modelThe coculture model was established using a Transwell apparatus, in which 1 × 105 TAM were seeded on the membrane of the Transwell insert and 5 × 105 organoids were seeded in the lower chamber of a 24-well plate for coculture.
2.4Luminex assayThe LabEX method was employed, and a Luminex 200 instrument was used to detect and quantify cytokines in the supernatant.
2.5Colony formation assay1000 cells were seeded into each well of a 6-well plate and incubated for 18 days at 37 °C. The cells were then fixed with 100 % methanol and stained with 0.1 % crystal violet.
2.6Flow cytometryOrganoids were incubated in a culture medium or medium supplemented with TAM and OSM with or without 5 μM Gem. After 48 h, the cells were collected using 0.25 % trypsin and 0.02 % EDTA, washed twice with PBS, stained with Annexin V-FITC/propidium iodide, and analyzed via flow cytometry.
2.7Cell viability assayAfter 48 hours of incubation, the medium was replaced with a medium containing different concentrations of gemcitabine. Cell viability was measured using the CellTiter-Glo assay after incubating at 37 °C for 96 h.
2.8RNA sequencingThe raw sequencing data were processed on the Majorbio I-Sanger Cloud Platform.
2.9Lentiviral transductionThe lentivirus-Flag-ID1 expression vector, lentivirus-sh-ID1 expression vector, lentivirus-Flag-Yap expression vector and lentivirus-sh-Yap expression vector were synthesized by ViGene BioSciences.
2.10ImmunofluorescenceCCLP-1 and Hucct-1 cells were fixed with 4 % paraformaldehyde and permeabilized with 0.02 % Triton X-100. Subsequently, the cells were incubated with an anti-Yap1 antibody overnight at 4 °C. Alexa Fluor 488-labeled goat anti-rabbit IgG was added to the cells, and the cells were incubated for 1 hour at room temperature.
2.11Western blotProtein separation was performed by SDS‒PAGE, and proteins were transferred onto a PVDF membrane. After blocking with 5 % skim milk, the membrane was incubated with primary antibodies overnight at 4 °C. The membrane was then incubated with secondary antibodies at room temperature for 1 hour.
2.12Co-IPCells were lysed, and the supernatant was collected after centrifugation for 30 min. The anti-Yap1 antibody was added to one portion of the lysate, and an anti-Importinα3 antibody was added to another portion. Subsequently, protein A/G agarose beads were washed with lysis buffer and centrifuged at 1000 × g for 3 minutes. The pretreated beads were added to both lysate portions, and the mixtures were incubated for 2–4 hours at 4 °C to couple the antibodies to the beads.
2.13Animal experimentsAll animal experiments were approved by the Ethics Committee of the Second Affiliated Hospital, Zhejiang University School of Medicine. For the tumor formation and drug resistance assays, 6-week-old nude mice were randomly grouped (n = 4), and each mouse was injected subcutaneously with 100 μl of cholangiocarcinoma cells (5 × 105) or cholangiocarcinoma cells (5 × 105) + tumor-associated macrophages (1 × 105).
2.14Statistical analysisAll statistical analyses were performed using GraphPad Prism software 7.0.
2.15Ethics statementThis study was conducted according to the Guidelines for the Laboratory Animal Use and Care Committee of the Ministry of Health, China and the Animal Research Ethics Committee of the Second Affiliated Hospital, Zhejiang University, School of Medicine (No. 2024–021).
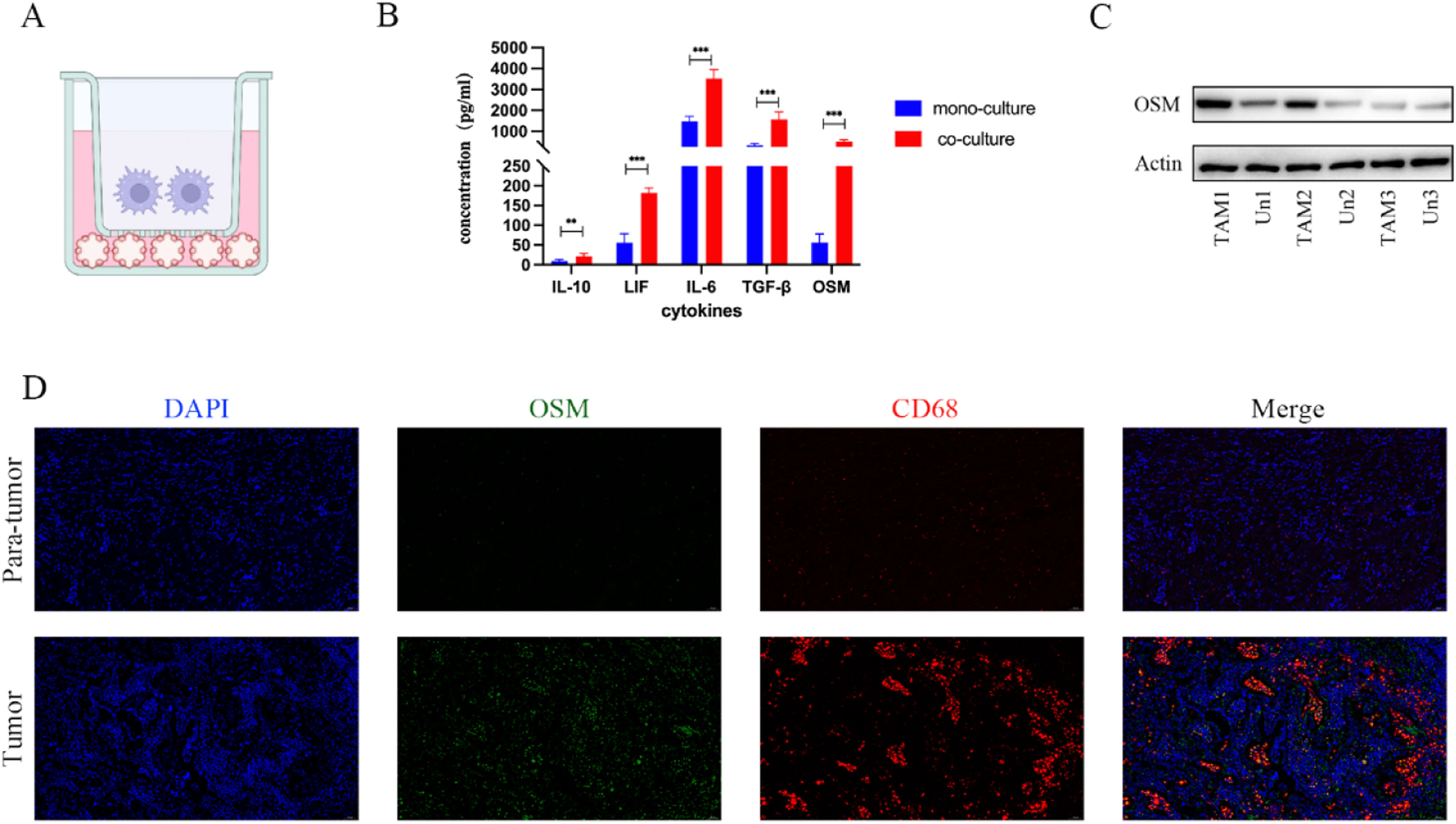
3Results3.1Tumor-associated macrophages produce oncostatin M in the tumor microenvironmentWe utilized a co-culture model of TAM and cholangiocarcinoma cells to explore their interactions in the tumor microenvironment. To investigate the changes in cytokine and chemokine induction by TAM in the tumor microenvironment, Luminex multiplex assays were employed. The introduction of TAM into the cell model led to significant increases in the levels of cytokines such as TGF-β, IL-6, and OSM, with OSM showing the most significant change (Fig. 1B & Supplementary: Fig. S1). To further confirm the source of OSM, we examined the protein levels of OSM in peripheral blood mononuclear cells (PBMC) from CCA patients and in TAM induced to differentiate from PBMC. We observed that the expression level of OSM was increased in TAM cultured in tumor cell-conditioned medium compared to TAM in the pre-induction state (Fig. 1C). The immunofluorescence staining results further confirmed that the expression of OSM in cholangiocarcinoma samples was localized around TAM in the microenvironment (Fig. 1D). These results indicate that TAM in the cholangiocarcinoma tumor microenvironment can secrete OSM.
 Schematic diagram of the co-culture model. (B) The Luminex assay results confirmed the increase in the OSM content in cell culture medium obtained from the coculture models. The data are shown as the mean ± SD of three independent experiments. ***p < 0.001. (C) The Western blot analysis results confirmed the changes in the protein level of OSM in TAM induced by the conditioned medium. (D) Representative images of OSM and CD68 expression in the tumor microenvironment obtained via multiplex immunofluorescence staining of cholangiocarcinoma samples and normal tissue samples. Scale bars: 50 μM.")
Tumor-associated macrophages produce oncostatin M in the tumor microenvironment. (A) Schematic diagram of the co-culture model. (B) The Luminex assay results confirmed the increase in the OSM content in cell culture medium obtained from the coculture models. The data are shown as the mean ± SD of three independent experiments. ***p < 0.001. (C) The Western blot analysis results confirmed the changes in the protein level of OSM in TAM induced by the conditioned medium. (D) Representative images of OSM and CD68 expression in the tumor microenvironment obtained via multiplex immunofluorescence staining of cholangiocarcinoma samples and normal tissue samples. Scale bars: 50 μM.
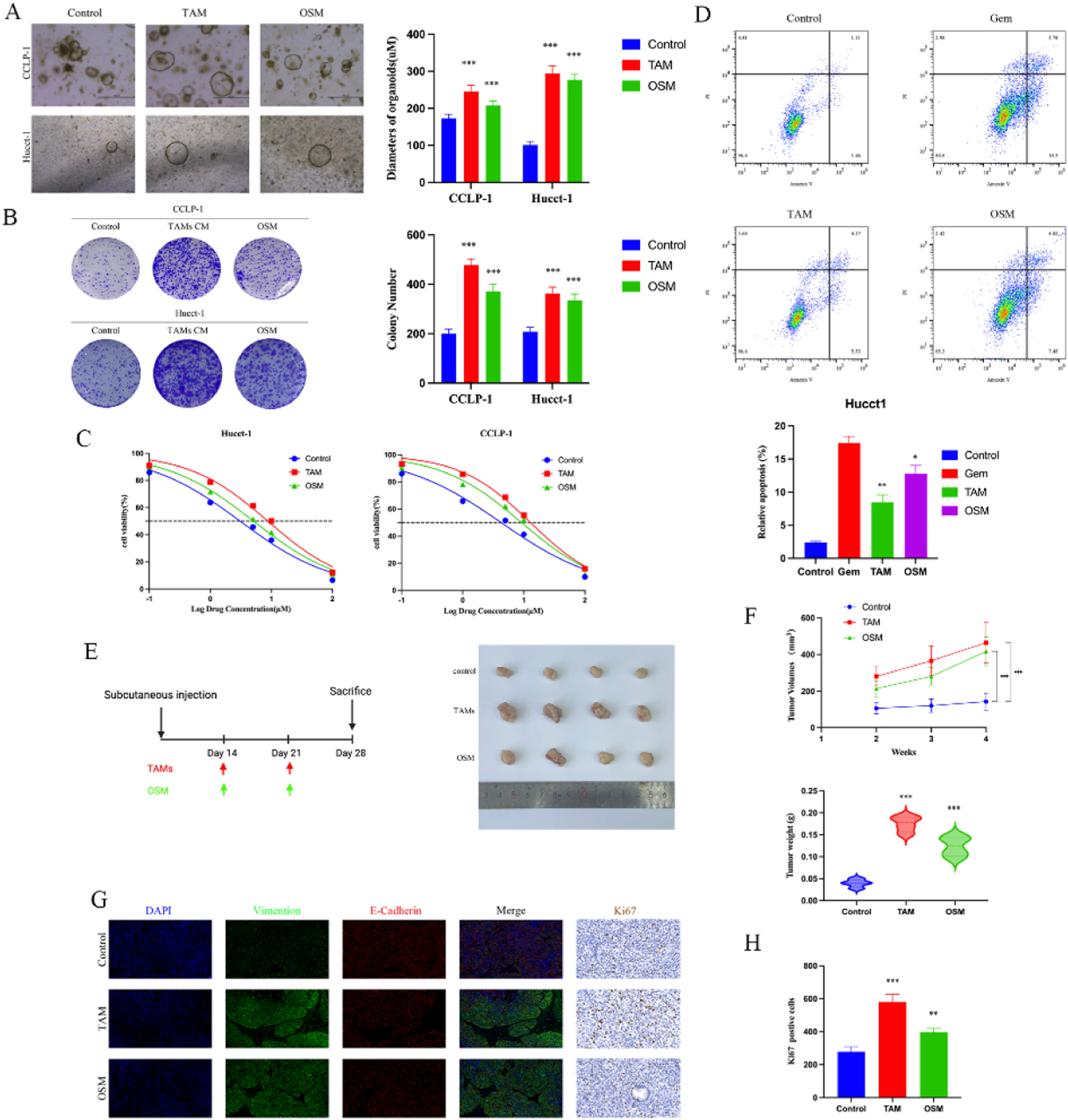
To further explore the impact of OSM on cholangiocarcinoma cells, we conducted cell proliferation and colony formation assays. We observed that the introduction of tumor-associated macrophages and 10 < ng/ml OSM significantly increased the diameter of cholangiocarcinoma organoids in the organoid models, with the TAM group showing the largest organoid diameter. Similar results were observed in the colony formation assays. We believe that the introduction of TAM leads to changes in the levels of various cytokines in the culture environment, which may be the key factor contributing to the increased proliferation and colony-forming ability of cholangiocarcinoma cells (Fig. 2 A-B). Additionally, we investigated whether TAM and OSM can protect CCA cells from the effect of Gem. As shown in Fig. 2C, the pro-apoptotic effect of Gem on tumor cells was decreased. FACS analysis of apoptosis revealed that treatment with TAM and OSM significantly reduced the pro-apoptotic effect of Gem on CCA cells compared to that in the control group, as shown in Fig. 2D.
 Representative bright-field images and diameters of long-term cultured CCA organoids i. Upper scale bars: 500 μM. Lower scale bars: 50 μM. The data are shown as the means±SD (n = 5 for each group). ***p < 0.001. (B) A colony formation assay and subsequent crystal violet staining were performed to determine colony numbers. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001. (C) Dose‒response curves for CCA organoids treated with Gem. The data are shown as the means±SD (n = 5 for each group). (D) CCA cells were incubated under different conditions as indicated in the presence or absence of 5 μM Gem for 48 h, and the percentage of apoptotic cells was determined via FACS. The data are shown as the mean ± SD of three independent experiments. (E) Schematic diagram of the design for the mouse xenograft experiments and Representative images showing tumors derived from transplanted organoids. (F) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 for each group). ***p < 0.001. (G) Representative images showing the expression of EMT-related proteins (E-cadherin and Vimentin) and Ki67 in tumors. (H) The positive of Ki67 in the three groups. The data are shown as the mean±SD (n = 3 for each group). ***p < 0.001.")
Biological effects of TAM and OSM on CCA. (A) Representative bright-field images and diameters of long-term cultured CCA organoids i. Upper scale bars: 500 μM. Lower scale bars: 50 μM. The data are shown as the means±SD (n = 5 for each group). ***p < 0.001. (B) A colony formation assay and subsequent crystal violet staining were performed to determine colony numbers. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001. (C) Dose‒response curves for CCA organoids treated with Gem. The data are shown as the means±SD (n = 5 for each group). (D) CCA cells were incubated under different conditions as indicated in the presence or absence of 5 μM Gem for 48 h, and the percentage of apoptotic cells was determined via FACS. The data are shown as the mean ± SD of three independent experiments. (E) Schematic diagram of the design for the mouse xenograft experiments and Representative images showing tumors derived from transplanted organoids. (F) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 for each group). ***p < 0.001. (G) Representative images showing the expression of EMT-related proteins (E-cadherin and Vimentin) and Ki67 in tumors. (H) The positive of Ki67 in the three groups. The data are shown as the mean±SD (n = 3 for each group). ***p < 0.001.
Then, we designed a mouse xenograft model to investigate the impact of TAM and OSM on the proliferation of cholangiocarcinoma cells in vivo. The results showed that the xenograft tumors composed of CCA cells cultured with TAM or OSM had significantly larger volumes and higher weights than did those in the control group (Fig. 2 E-F). The immunofluorescence staining results demonstrated that the presence of TAM and OSM promoted epithelial-mesenchymal transition (EMT) in cholangiocarcinoma cells, as evidenced by the significant increase in Vimentin expression (Fig. 2G). Similar results were observed for immunohistochemical staining of Ki67 (Fig. 2H). These findings indicate that TAM and OSM in the cholangiocarcinoma microenvironment promote the proliferation and clonogenicity of CCA cells in vitro and enhance tumor growth in vivo.
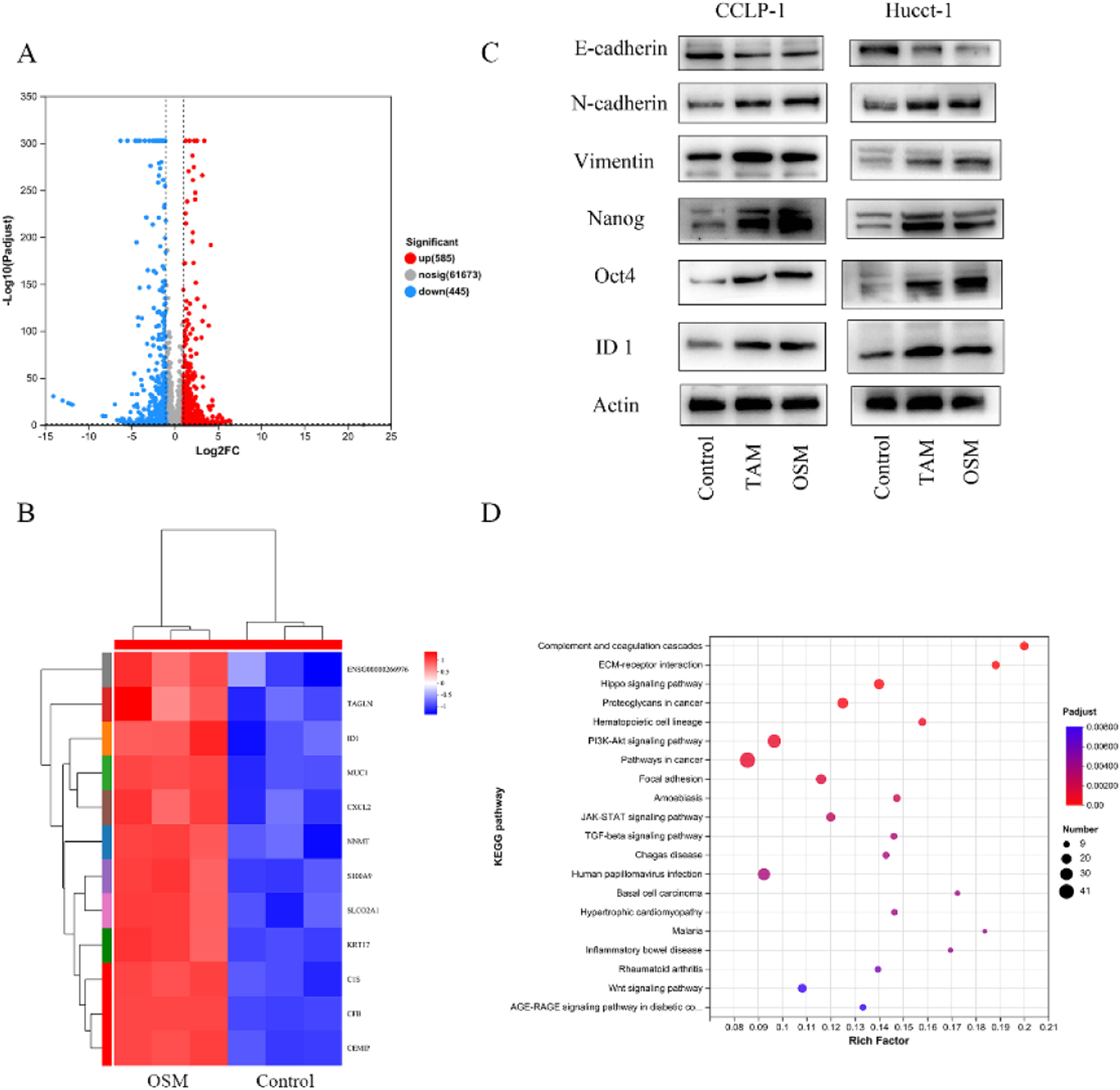
3.3Transcriptome analysis to explain the biological effects of OSM on cholangiocarcinomaTo further investigate the targets of OSM in CCA cells, we compared the transcriptomes of OSM-treated CCA organoids with those of control organoids and identified 1030 differentially expressed genes. Among them, 585 genes were upregulated and 445 genes were downregulated after OSM treatment (Fig. 4A). TAGLN (transgelin), ID1 (inhibitor of DNA binding 1), MUC1 (mucin 1, cell surface associated), CXCL12 (C-X-C motif chemokine ligand 12), NNMT (nicotinamide N-methyltransferase), S100A9 (S100 calcium binding protein A9), and other genes were highly expressed in the OSM-treated organoids (Fig. 3B). Analysis of the differentially expressed genes revealed that the ID1 gene is associated with epithelial-mesenchymal transition (EMT). To confirm whether OSM promotes EMT and stemness by upregulating ID1 expression, Western blot analysis was performed, revealing that OSM-treated tumor cells exhibited decreased expression of E-Cadherin and increased expression of N-Cadherin and Vimentin. Additionally, the expression of the tumor stemness markers Nanog and Oct4 was significantly increased (Fig. 3C). Subsequently, the results of gene set enrichment analysis revealed the enrichment of pathways such as "Complement and coagulation cascades," "ECM-receptor interaction," "Hippo signaling pathway," and "Proteoglycans in cancer" in OSM-treated organoids (Fig. 3D).
 on cholangiocarcinoma cells. (A) Volcano plot showing the differentially expressed genes (DEGs) between control and OSM-treated cancer organoids. Differential gene expression was statistically analyzed by the DESeq method and defined by two criteria: an absolute log2 (fold change) value of >1 and a P value of < 0.05. A total of 585 genes were upregulated and 445 genes were downregulated in the OSM-treated cancer organoids. (B) Heatmap showing the differentially expressed genes of Hucct-1. Differential gene expression was statistically analyzed by the DESeq method and defined by two criteria: an absolute log2 (fold change) value of >1 and a P value of < 0.05. (C) Western blot analysis of EMT-related genes (E-cadherin, N-cadherin and vimentin) and stem cell markers (Nanog and Oct-4) in CCA cells. (D) Gene set enrichment analysis of enriched Kyoto Encyclopedia of Genes and Genomes pathways.")
Transcriptome analysis of the effect of oncostatin M (OSM) on cholangiocarcinoma cells. (A) Volcano plot showing the differentially expressed genes (DEGs) between control and OSM-treated cancer organoids. Differential gene expression was statistically analyzed by the DESeq method and defined by two criteria: an absolute log2 (fold change) value of >1 and a P value of < 0.05. A total of 585 genes were upregulated and 445 genes were downregulated in the OSM-treated cancer organoids. (B) Heatmap showing the differentially expressed genes of Hucct-1. Differential gene expression was statistically analyzed by the DESeq method and defined by two criteria: an absolute log2 (fold change) value of >1 and a P value of < 0.05. (C) Western blot analysis of EMT-related genes (E-cadherin, N-cadherin and vimentin) and stem cell markers (Nanog and Oct-4) in CCA cells. (D) Gene set enrichment analysis of enriched Kyoto Encyclopedia of Genes and Genomes pathways.
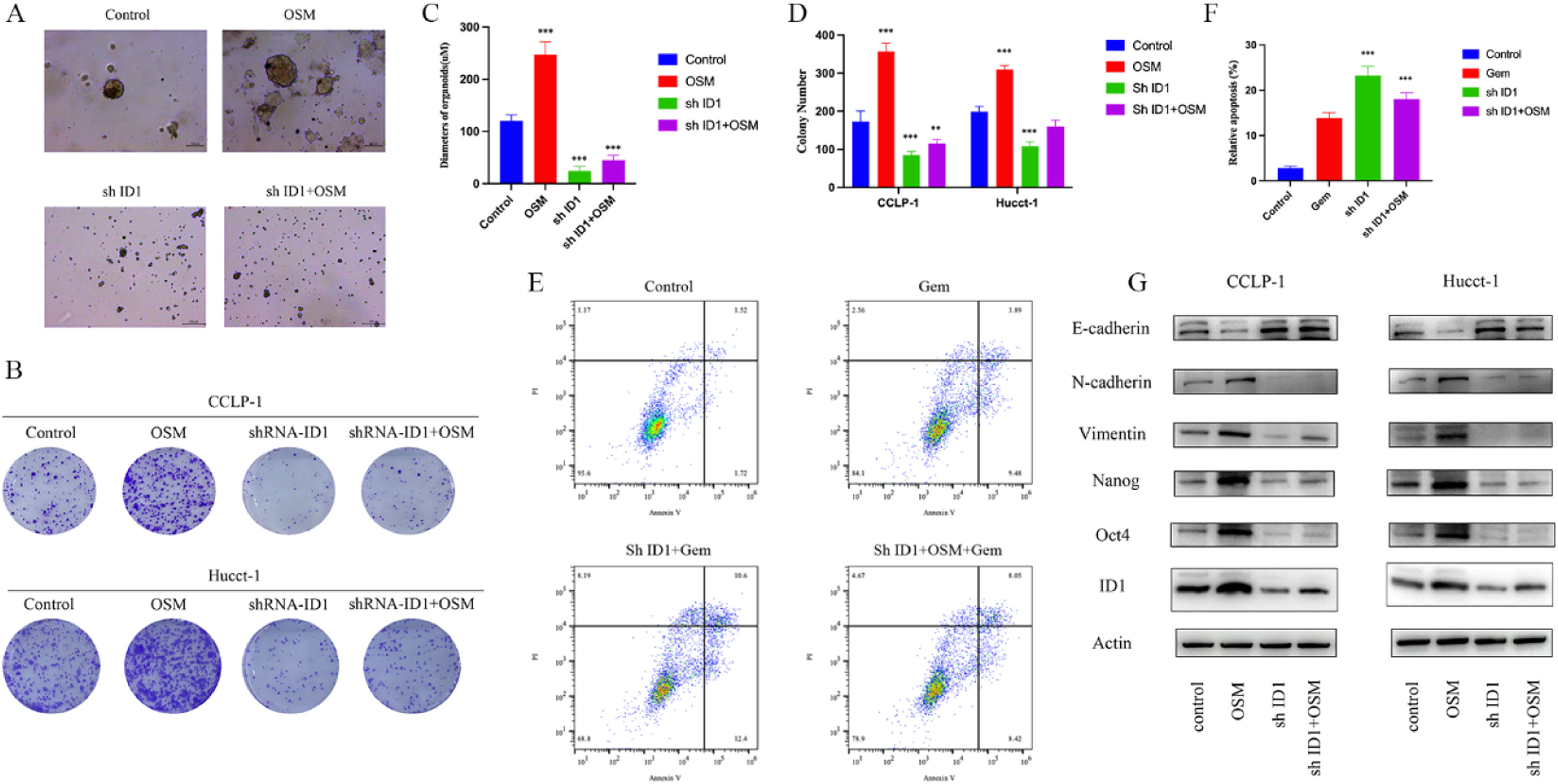
To verify whether ID1 is involved in the OSM-mediated acceleration of CCA cell proliferation and confirm its protective effect against Gem-induced apoptosis, we generated cholangiocarcinoma cells with ID1 knockdown using lentivirus. We observed that, under long-term culture in organoid models, cholangiocarcinoma cells with decreased expression of the transcription factor ID1 were unable to form extensive three-dimensional cellular structures. Even after the addition of OSM to the culture medium, the diameter of the organoids was significantly smaller than that of the organoids in the control group (Fig. 4A, C). The colony formation assay results showed a similar trend. Additionally, ShID1 transduction combined with OSM supplementation promoted the formation of cell colonies compared to that in the group with only the knockdown of ID1 (Fig. 4B, D). Furthermore, we validated the role of ID1 in suppressing Gem-mediated apoptosis in cholangiocarcinoma cells. We found that knocking down ID1 significantly increased the percentage of cholangiocarcinoma cells with Gem-induced apoptosis (Fig. 4E, F). Western blot analysis demonstrated decreases in the expression of N-Cadherin and Vimentin in cholangiocarcinoma cells with ID1 knockdown, as well as significant reductions in the expression of the tumor stem cell markers Nanog and Oct4 (Fig. 4G). Thus, these in vitro experiments showed that OSM induces EMT and increases tumor stemness through ID1, thereby promoting tumor cell proliferation and resistance to Gem.
 Representative bright-field images and diameters of CCA organoids in the four groups. Scale bars: 100 μM. The data are shown as the means±SD (n = 5 for each group). ***p < 0.001. (C-D) A colony formation assay and subsequent crystal violet staining were performed to determine colony numbers. The data are shown as the means±SD (n = 3 for each group). **p < 0.01, ***p < 0.001. (E-F) CCA cells were incubated under different conditions as indicated in the presence or absence of 5 μM Gem for 48 h, and the percentage of apoptotic cells was analyzed via FACS. The data are shown as the mean ± SD of three independent experiments. (G) Western blot analysis of EMT-related genes (E-cadherin, N-cadherin and vimentin), stem cell markers (Nanog and Oct-4), and ID 1 in CCA.")
Biological effect of OSM on CCA and the related mechanism. (A-B) Representative bright-field images and diameters of CCA organoids in the four groups. Scale bars: 100 μM. The data are shown as the means±SD (n = 5 for each group). ***p < 0.001. (C-D) A colony formation assay and subsequent crystal violet staining were performed to determine colony numbers. The data are shown as the means±SD (n = 3 for each group). **p < 0.01, ***p < 0.001. (E-F) CCA cells were incubated under different conditions as indicated in the presence or absence of 5 μM Gem for 48 h, and the percentage of apoptotic cells was analyzed via FACS. The data are shown as the mean ± SD of three independent experiments. (G) Western blot analysis of EMT-related genes (E-cadherin, N-cadherin and vimentin), stem cell markers (Nanog and Oct-4), and ID 1 in CCA.
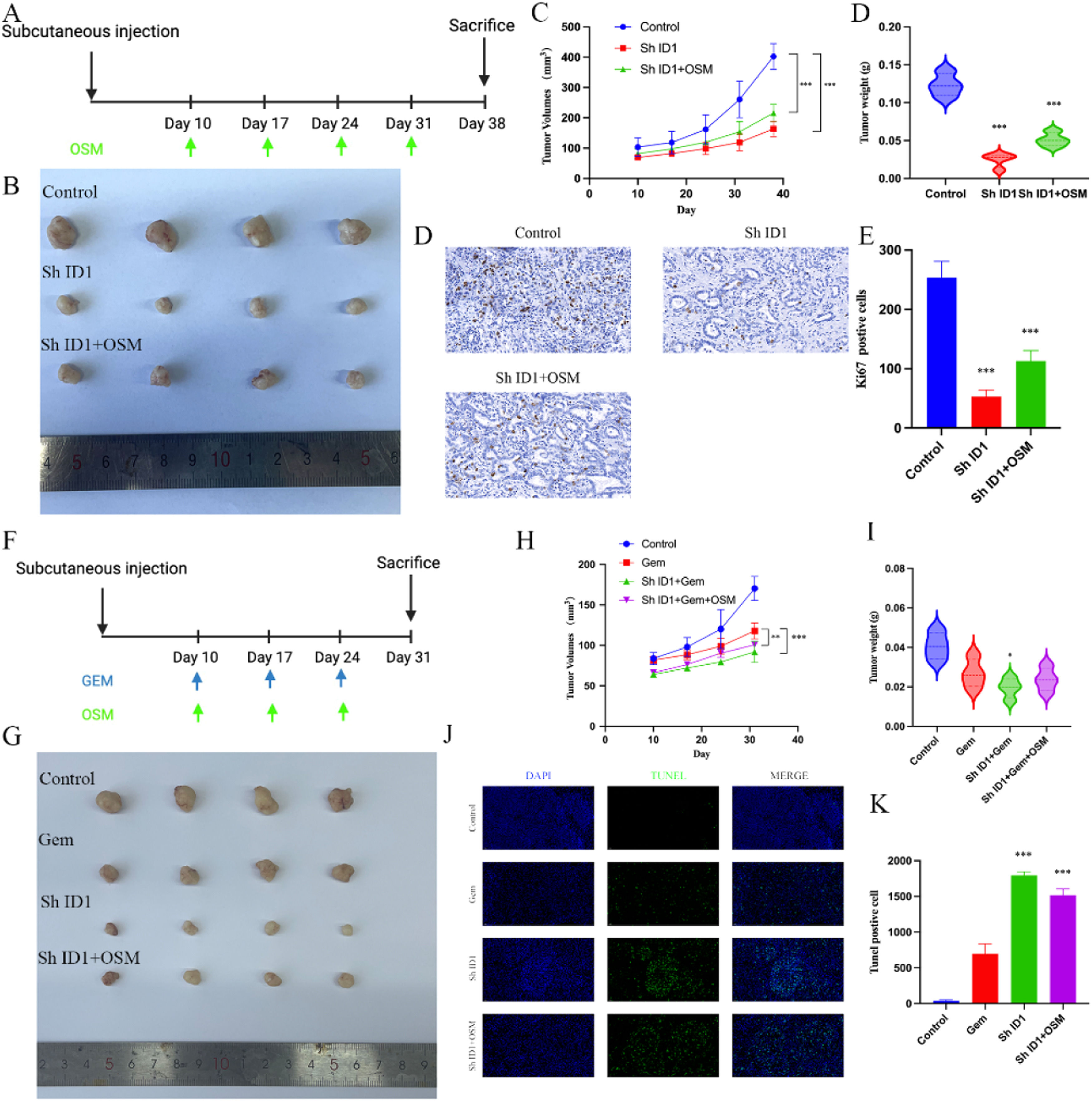
Due to the crucial role of ID1 observed in the above in vitro cell experiments, further exploration of the role of ID1 at the organism level became the primary focus of the study. After successfully establishing cells with stable ID1 knockdown, we implanted CCA organoids into nude mice to establish a subcutaneous xenograft model and simultaneously recorded the tumor weight and volume. The experimental design for the xenograft tumor experiment is shown in Fig. 5A. The results indicated significant inhibition of xenograft tumor growth after knocking down the transcription factor ID1 (Fig. 5 B-C). Even upon stimulation with OSM after tumor formation, the tumor volume and weight were lower than those in the control group. Simultaneously, we performed immunohistochemical staining for Ki67 to assess the proliferation of tumor cells. The ID1 knockdown group exhibited fewer Ki67-positive cells than did the other two groups (Fig. 5 D-E). Both the in vivo and in vitro experimental results confirmed the important role of ID1 in the process by which OSM promotes tumor formation. Additionally, we explored whether ID1 has a suppressive effect against Gem-induced apoptosis. Mice in all groups except for the blank control group received regularly scheduled injections of 50 mg/kg gemcitabine. The results indicated that knocking down ID1 increased the susceptibility of tumor cells to the proapoptotic effects of Gem. The tumor volume and weight were significantly lower in the Gem+ID1 knockdown group than in the Gem group, and the number of TUNEL-positive cells was highest in the Gem+ID1 knockdown group (Fig. 5 G-K).
 Schematic diagram of tumor growth in the CCA xenograft assay. (B) Representative images of xenograft tumors from three different groups are shown. (C) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 mice for each group). ***p < 0.001. (D) Representative immunohistochemical staining of Ki67 in the three groups. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001. (E) Schematic diagram of chemoresistance in the CCA xenograft assay. (F) Representative images of xenograft tumors from four different groups are shown. (G) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 mice for each group). *p < 0.05, **p < 0.01, ***p < 0.001. (H) Representative images of TUNEL-positive cells in the four groups. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001.")
The macrophage-OSM-ID1 feedback loop promotes CCA growth and gemcitabine resistance in vivo. (A) Schematic diagram of tumor growth in the CCA xenograft assay. (B) Representative images of xenograft tumors from three different groups are shown. (C) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 mice for each group). ***p < 0.001. (D) Representative immunohistochemical staining of Ki67 in the three groups. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001. (E) Schematic diagram of chemoresistance in the CCA xenograft assay. (F) Representative images of xenograft tumors from four different groups are shown. (G) The tumor volume, tumor growth curve and tumor weight are shown. The data are shown as the means±SD (n = 4 mice for each group). *p < 0.05, **p < 0.01, ***p < 0.001. (H) Representative images of TUNEL-positive cells in the four groups. The data are shown as the means±SD (n = 3 for each group). ***p < 0.001.
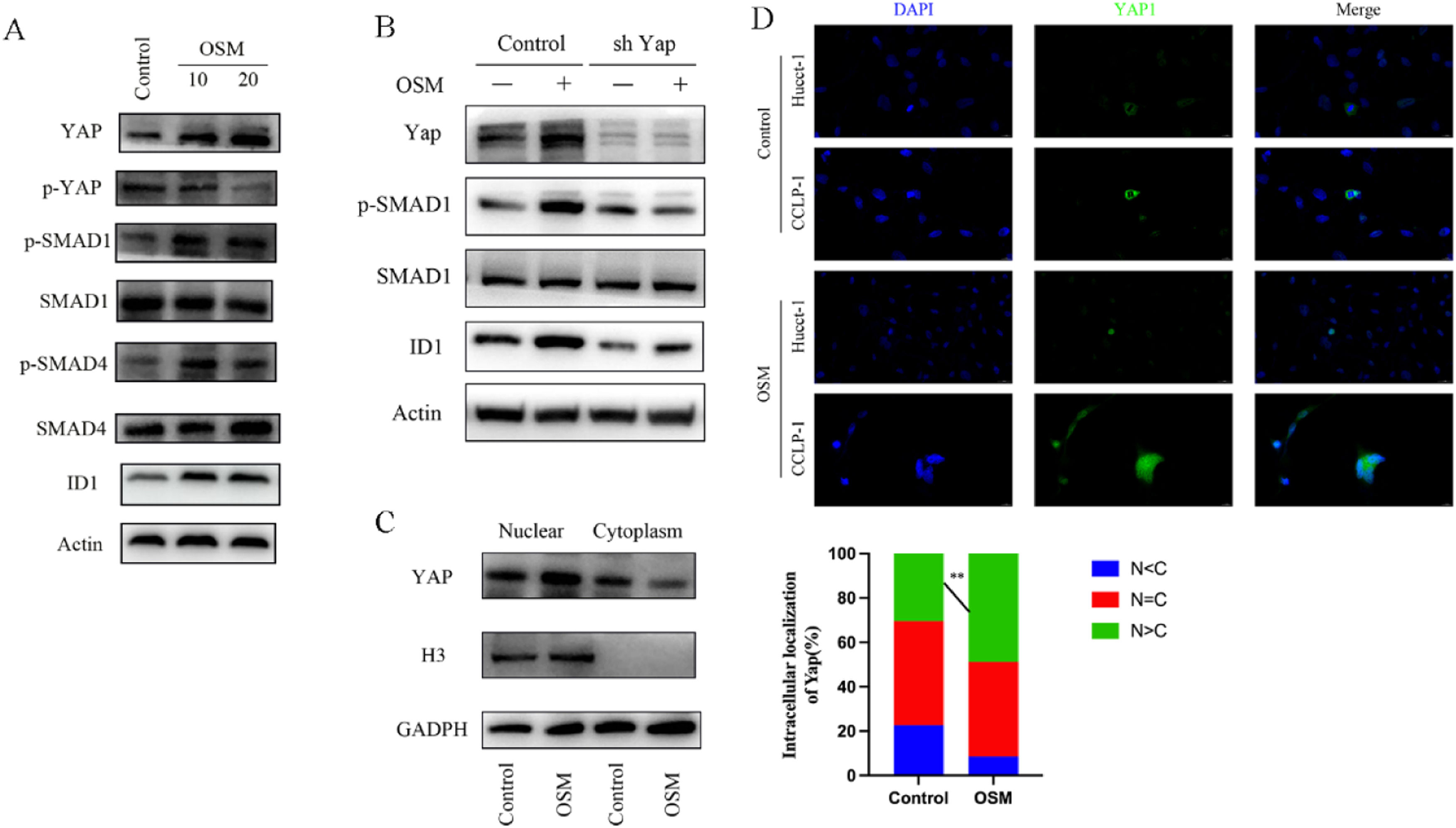
Based on our transcriptome enrichment analysis, we hypothesized that the Hippo-YAP pathway may be involved in ID1 regulation. In the context of the Hippo signaling pathway, the regulatory effect of YAP on ID1 expression is not fully understood. Previous studies have confirmed that SMAD1, when activated through phosphorylation, forms complexes with other SMAD proteins, regulating the expression of the transcription factor ID1 [24,25]. Studies have suggested the occurrence of crosstalk between the Hippo–YAP and TGF-β/SMAD signaling pathways, with YAP recruited to interact with phosphorylated SMAD1 in the nucleus [26,27]. Moreover, it has been confirmed that IL-6 promotes the migration and invasion of tumor cells by regulating Yap nuclear translocation, and OSM is one of the members of the IL-6 cytokine family [28]. Thus, we speculated that YAP and phosphorylated SMAD1 may be involved in regulating the expression of the transcription factor ID1. After treatment of cholangiocarcinoma cells with 10 < ng/ml and 20 < ng/ml OSM for 7 days, Western blot analysis revealed significant increases in the levels of YAP and phosphorylated SMAD1 in CCA cells (Fig. 6A-B). However, there was no significant difference between phosphorylated SMAD2 and SMAD3 expression (Supplementary: Fig. S2). Subsequently, we separately extracted nuclear and cytoplasmic proteins, verified the purity of the nuclear proteins through analysis of histone H3, and confirmed that OSM stimulation promoted the nuclear translocation of YAP, leading to a significant increase in the abundance of nuclear YAP (Fig. 6C). This conclusion was further confirmed through immunofluorescence staining of CCLP-1 and Hucct-1 cells (Fig. 6D).
 OSM activates ID1 through YAP-SMAD1 signaling. CCA cells were treated with OSM (10 ng/ml or 20 ng/ml) for 7 days, after which protein expression was analyzed via Western blotting. (C) Western blotting showing YAP expression in the nucleus and cytoplasm. (D) Representative images of CCA cells stimulated with OSM (10 ng/ml) for 7 days and stained for YAP.")
OSM activates ID1 in CCA cells through YAP nuclear enrichment. (A-B) OSM activates ID1 through YAP-SMAD1 signaling. CCA cells were treated with OSM (10 ng/ml or 20 ng/ml) for 7 days, after which protein expression was analyzed via Western blotting. (C) Western blotting showing YAP expression in the nucleus and cytoplasm. (D) Representative images of CCA cells stimulated with OSM (10 ng/ml) for 7 days and stained for YAP.
Furthermore, we found that even under OSM stimulation, the level of phosphorylated SMAD1 in the sh Yap group was almost identical to that in the control group. In further exploring the mechanisms related to YAP nuclear translocation, we discovered that the shuttling of STAT3 from the cytoplasm to the nucleus is mediated by Importinα3. OSM has been confirmed to promote STAT3 phosphorylation and nuclear translocation. Therefore, we hypothesized that YAP nuclear translocation might depend on Importinα3. We detected endogenous Importinα3 in the Flag-tagged YAP immunoprecipitates in transduced CCA cells (Supplementary: Fig. S3). Based on these results, we propose that OSM stimulation in the cholangiocarcinoma microenvironment promotes YAP nuclear translocation through Importinα3.
4DiscussionTumor organoids have broad applications in the fields of basic cancer research, new drugs and precision therapy. However, constructing the tumor microenvironment in which to study mechanisms related to tumor heterogeneity, cancer progression, drug resistance, etc., poses a considerable challenge in cancer research. In this study, monocytes were obtained from peripheral blood and differentiated into TAM. Subsequently, these TAM were cocultured with cholangiocarcinoma organoids. The results indicated that TAM can secrete OSM, promoting the proliferation and chemoresistance of CCA.
Many studies using conditioned medium from cholangiocarcinoma cell lines and macrophage lines have shown that TAM increases the proliferation, invasion, and colony formation of CCA in vitro [29]. In the present study, monocytes were induced to differentiate into TAM using cytokines and conditioned medium. These TAM were then cocultured with tumor cells in an organoid model to explore the interaction between two cell types. We believe that a 3D model involving direct contact between tumor organoids and immune cells is a direction for further development, as this model would provide a more authentic representation of the tumor microenvironment for future cancer research applications.
Through coculturing TAM with CCA cells, we observed changes in the levels of many cytokines and chemokines in the culture environment. To validate this hypothesis, we screened for significantly altered cytokines through Luminex assays and confirmed the source of OSM. OSM is a member of the interleukin-6 family of cytokines and is a multifunctional factor involved in tumor progression, increased tumor stemness, and the occurrence of drug resistance [30]. Furthermore, due to its impact on various tumor-related signaling pathways, OSM is considered an effective therapeutic target as an antiproliferative factor in certain cancers.
Studies have reported that interactions between TANs and TAM promote ICC progression through the OSM/IL-11/STAT3 signaling pathway in cholangiocarcinoma [31]. Our study revealed that the stimulation with the factor OSM increased tumor cell proliferation and gemcitabine resistance. We believe that TAM may also secrete other cytokines, which cooperate with OSM to play a role in promoting tumorigenesis.
STAT3 is the primary downstream signaling molecule of OSM. Previous studies have indicated that crosstalk occurs between OSM/STAT3 and the TGF-β/SMAD signaling pathway, influencing EMT and the acquisition of cancer stem cells (CSCs) characteristics [32]. CSCs are a population of cells present in tumors and are characterized by their promotion of EMT, self-renewal, and chemoresistance. Transcriptome results revealed upregulation of the transcription factor of ID1 in CCA stimulated long-term with OSM. Previous research has confirmed the involvement of ID family proteins in regulating cancer stem cells, with several studies demonstrating the dysregulation of ID1 in cancer, in which it promotes tumor cell growth, survival, invasion, migration, and angiogenesis [33,34]. Our results indicated that OSM promoted EMT and increased tumor stemness by upregulating ID1 expression. Therefore, we propose that OSM accelerates tumor cell proliferation and gemcitabine resistance primarily through upregulation of the transcription factor ID1.
The results of transcriptome showed that these genes induced by OSM on CCA cells are correlated with the Hippo-Yap pathway. However, the current understanding of the relationship between Yap and ID1 is incomplete. Our Studies have confirmed that phosphorylated SMAD1 forms complexes with other SMAD proteins, which then act as transcription factors to regulate the expression of the transcription factor ID1. Evidence suggests that crosstalk occurs between the Hippo/YAP and TGF-β/SMAD signaling pathways, with Yap interacting with SMAD1. Importantly, the nuclear transport protein Importin3, which is required for STAT3 nuclear translocation, is also necessary for YAP nuclear import.
5ConclusionsIn conclusion, our study establishes that OSM secreted by TAM upregulates the expression of YAP, leading to the upregulation of ID1, which subsequently promotes EMT and increases tumor stemness. Consequently, these promoting effects contribute to tumor proliferation and the emergence of drug resistance. These findings support the exploration of TAM and OSM as potential therapeutic targets for CCA.
FundingThis work was supported by the Major science and technology program of Fujian Province under Grant [2021YZ036017].
Author contributionsYHG: Conceptualization, Methodology, Software, Investigation, Formal analysis, Writing -Original Draft, SDM: Conceptualization, Methodology, Software, Investigation, Formal analysis, Writing-Original Draft, YJ: Resources, QL: Resources, YHW: Resources, XXZ: Resources, J.T.L.: Writing - Review & Editing, Visualization, Supervision, Project administration, Funding acquisition.
Availability of data and materialsThe datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.



