


Anthracyclines are cytostatic antibiotics discovered almost half a century ago exerting their action through inhibition of topoisomerase II. The two most representative drugs are doxorubicin and daunorubicin and they have been proven as useful antineoplastics and are widely prescribed in daily oncology practice; unfortunately, cardiotoxicity has been a limiting factor when it comes to their use.
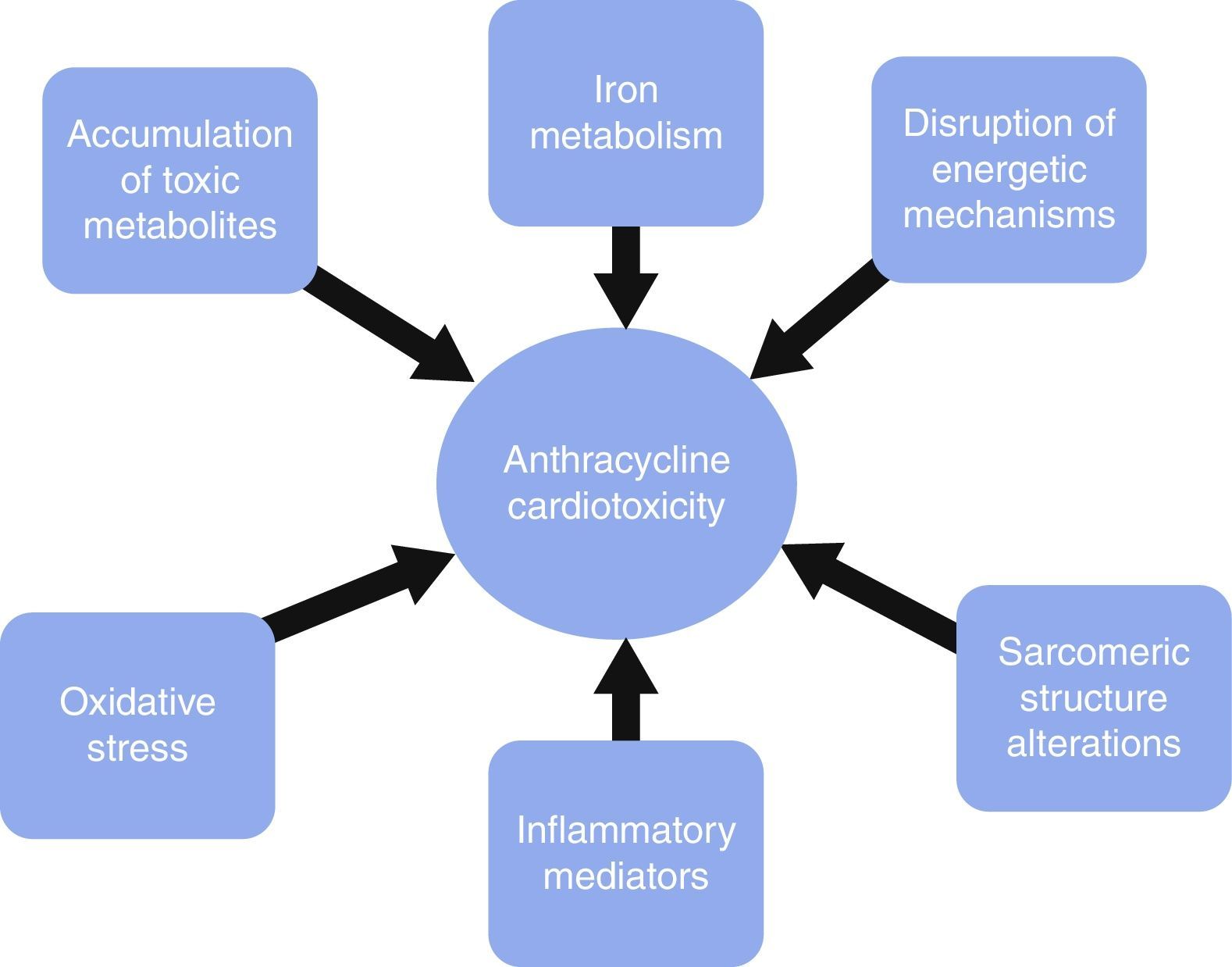
Diverse mechanisms have been involved in anthracycline cardiotoxicity, none of which are capable of causing the whole clinical picture by itself. Traditionally, reactive oxygen species (ROS) have received more attention, although recently basic research has proven other factors to be as important as ROS. These factors mainly involve sarcomeric structure disruption, toxic accumulation of metabolites, iron metabolism, energetic alterations and inflammation.
The role of genetics has been studied by some groups, although a clear genotype-response relationship is yet to be elucidated.
With the improved survival from different oncologic diseases we are witnessing more cases of chemotherapy-induced cardiotoxicity and the advent of new anticancer drugs poses several challenges for the cardiologist, highlighting the importance of a deep knowledge of the main mechanisms inducing this toxicity.
Hace casi medio siglo se descubrieron las antraciclinas; estas son antibióticos citostáticos inhibidores de la topoisomerasa ii. Los 2 fármacos más representativos de este grupo son la doxorrubicina y la daunorrubicina. Estos fármacos han demostrado ser eficaces antineoplásicos y han sido ampliamente utilizados en la práctica oncológica. Desafortunadamente, la cardiotoxicidad sigue siendo un elemento limitante para su uso.
Los mecanismos mediante los cuales estos fármacos ocasionan cardiotoxicidad son múltiples pero ninguno de ellos de forma individual es capaz de explicar el cuadro clínico por completo. Casi siempre se ha considerado que la formación de especies reactivas de oxígeno era responsable de gran parte de la toxicidad, sin embargo la experimentación básica reciente ha demostrado que hay otros factores, entre los que destacan las alteraciones en la estructura sarcomérica, la acumulación de metabolitos tóxicos, las alteraciones del metabolismo del hierro o de los mecanismos energéticos, y la liberación de mediadores de inflamación.
Por otra parte, diversos grupos han investigado la intervención que la genética podría tener en el desarrollo de esta enfermedad, si bien no se puede definir aún una clara correlación genotipo-respuesta.
Con el aumento de la supervivencia por el tratamiento de diversas enfermedades oncológicas, se están detectando más casos de cardiotoxicidad mediada por quimioterapia; y con la aparición de nuevos fármacos quimioterápicos se añaden nuevos retos, con lo que se demuestra la importancia del estudio profundo de los mecanismos causales.
Anthracyclines are cytostatic antibiotics discovered almost half a century ago that exert their action through inhibition of topoisomerase II.1,2 The two most representative drugs of this group are doxorubicin (active against some hematologic cancers and solid tumors)3 and daunorubicin (used mainly in acute hematologic cancer),4 although other drugs have been developed within this family (e.g. epirubicin, idarubicin and mitoxantrone).
These drugs have been proven as useful antineoplastics and are amongst the most widely prescribed oncologic medication. Unfortunately, cardiotoxicity secondary to anthracyclines still remains a limiting factor when used,5 being generally related to dose–response (although there are several reports of cardiotoxicity associated with lower doses, probably secondary to individual susceptibility)6,7and manifesting mainly as heart failure usually within a year after completion of treatment, although it is widely accepted that it can manifest itself several years after treatment.
Recently, groups have been created in Europe and the United States for the study of this toxicity, highlighting its importance.8,9
Scope of the problemChemotherapy-induced cardiotoxicity is usually classified in two groups according to the cellular damage induced by these drugs. Type I cardiotoxicity implies cellular death (either via necrosis or apoptosis) and thus, is not reversible, whereas type II cardiotoxicity is caused by cellular dysfunction (not death) and is usually described as reversible. Type I is characteristic of anthracycline damage, while type II is more frequent with monoclonal antibodies (namely, trastuzumab).8
Gianni et al.5 reported some of the several limitations that have emerged in terms of describing the current situation of anthracycline-mediated cardiotoxicity; these problems come from a lack of uniformity in reporting or detecting events in these patients and the different forms of presentation (acute vs. delayed, reversible vs. irreversible). At the same time, the wide difference in the treated populations (adults vs. children) makes it difficult to unify criteria. Nevertheless, recent statistics regarding survival of oncologic patients give us an overview of the problem.
At the moment, in the US there are more than 300,000 survivors of childhood cancer, of whom 50% could potentially have been treated with anthracyclines,10 and the probability of accumulated death due to cardiac cause (including sudden death likely to be cardiac in origin) is greater than the observed in control groups.11,12
Likewise, survival of breast cancer (one of the clinical scenarios where these drugs have been used extensively) has increased dramatically in the last decades, translating into approximately two million women in the US with high likelihood of previous anthracycline exposure.13
Despite several efforts to diminish this adverse effect (mainly by reducing the cumulative dose under 400–450mg/m2 for doxorubicin) and considering the previously mentioned caveats, studies estimate heart failure rates between 5 and 10%5,8 with the modern regimens of treatment, depending on age, cumulative dose, individual susceptibility or even past medical history (e.g. hypertension, coronary heart disease, etc.), among other factors.
Mechanisms of cardiotoxicityWith all the aforementioned, it is not surprising that considerable controversy has risen with respect to the mechanisms involved in this toxicity. Anthracycline-induced cardiotoxicity seems to be a multi-step condition, with several pathways involved. Many research groups have tried to elucidate the processes associated; of these, we will focus in this review on those that have been most extensively studied.
It is important to acknowledge the fact that these pathways are not necessarily independent from each other or exclusive, as each of them may play a role in causing cardiotoxicity via different mechanisms, by themselves or in cooperation with other pathways. Fig. 1 depicts some of the mechanisms generating cardiotoxicity.

We will not review in the present work the rare, but existent, acute presentation of anthracycline toxicity or the involvement of the pericardium.
Oxidative stressOxidative stress is the most widely studied mechanism and it involves a highly complex pathophysiology, which is a matter of permanent debate.
The myocardium is extremely prone to oxidative damage and this is, at least in part, due to its lower levels of catalase activity and superoxide dismutase (an enzyme that catalyses the dismutation of superoxide, one of the main reactive oxygen species [ROS])14 compared with other tissues.
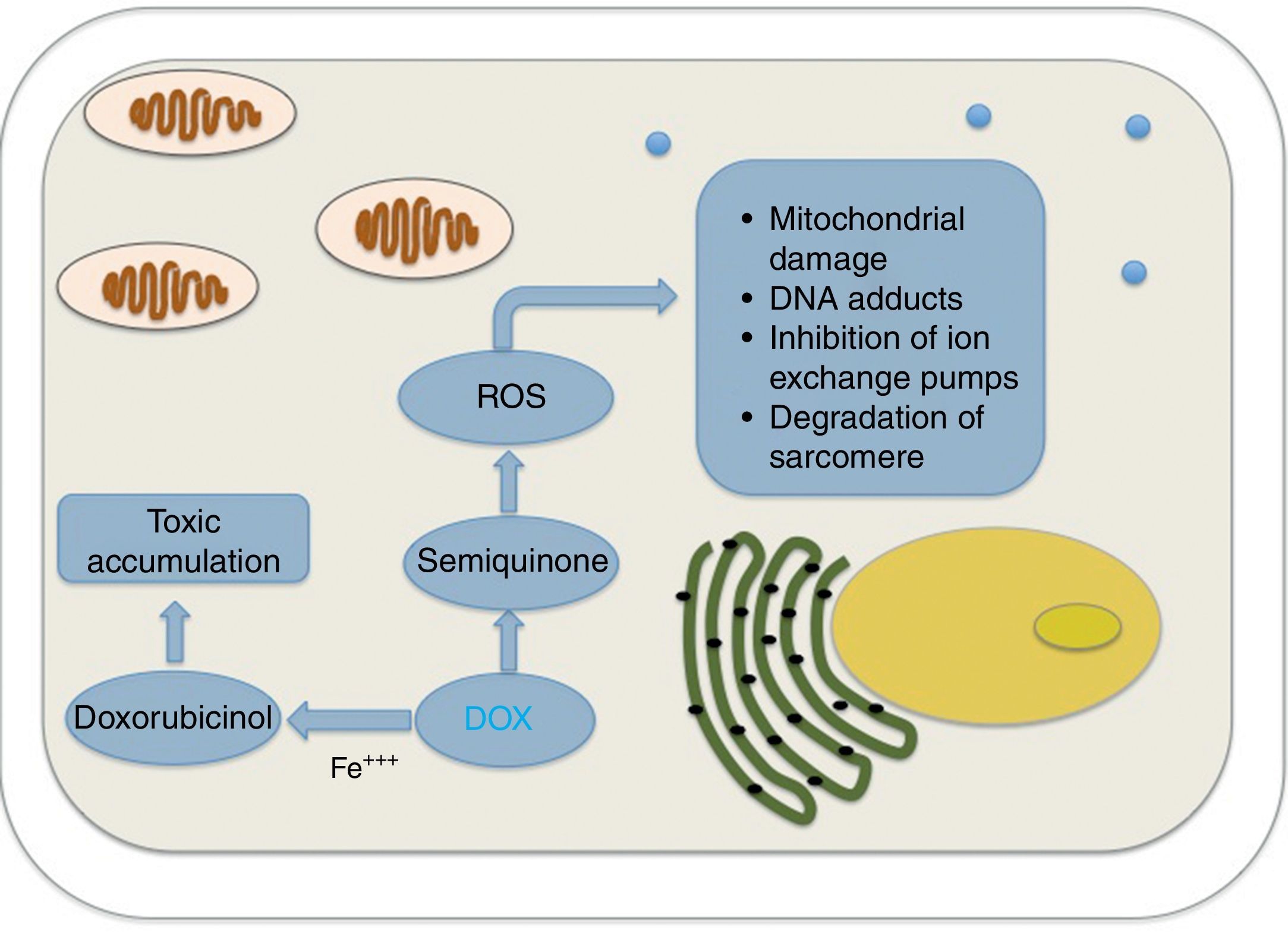
The metabolism of anthracyclines involves the reduction of the quinone fraction of its formula to semiquinone, which can rapidly transfer its unpaired electron to an electron acceptor (usually molecular oxygen), returning to its original quinone form, therefore completing the redox cycle and leading to the formation of more ROS.
Other mechanisms involved include the formation of DNA adducts by semiquinone or the generation of superoxide anions by anthracycline metabolism, with subsequent cellular damage by degradation of the sarcomere, mitochondrial dysfunction and DNA damage.15,16
A recent paper by Octavia et al.17elegantly describes the different pathways by which anthracyclines cause ROS, including mitochondrial, nitric oxide synthase (NOS), and nicotinamide adenine dinucleotide phosphate (NADPH) pathways.
Even though the evidence for myocardial damage induced by ROS is vast, oxidative stress as the sole cause of cardiotoxicity is increasingly questioned and more research is dedicated to alternative pathways. Part of the reason for this came from earlier studies that failed to prove the protective role of certain antioxidants in the long term,5 especially when interventions that resembled the ones used in clinical practice were tested.15 It is noteworthy that some of the drugs that might have a protective role (e.g. dexrazoxane and carvedilol) probably exert their defensive mechanism through alternative molecular routes, besides their antioxidant activity. Fig. 2 shows the relationships between DOX, doxorubicinol and ROS in cardiotoxicity.
Accumulation of toxic metabolites
Anthracyclines metabolism generates toxic molecules at the myocardium level through a reduction of their carbonyl group (producing doxorubicinol in case of doxorubicin, being up to 50 times more potent than the original compound, or daunorubinicol and idarubicinol in case of daunorubicin and idarubicin, respectively),17 capable of inhibiting the ion exchange pumps of calcium and sodium, even at the mitochondrial level, causing an imbalance in the energetics of the myocardium and a diminished systolic function.18 Moreover, these secondary alcohol metabolites are more difficult to eliminate from the cardiomyocyte than the parent drug, accumulating inside them.19
All the anthracyclines have shown to have similar mechanisms of bioactivation, making them toxic from the cardiac standpoint,2 but the role of toxic metabolites has been challenged by the fact that both daunorubicin and idarubicin generate higher plasma levels of alcohol metabolites compared with doxorubicin15 and they tend to generate less cardiotoxicity.
A more detailed description of the molecular damage induced by each drug is beyond the scope of this paper.
Iron metabolismIron has a major role in cell metabolism and is under control by several regulatory systems.20 Its functions go far beyond the oxygen transport complex, being part of different processes like bioenergetics, enzymatic coordination or even immune system physiology.
Traditionally, it has been considered that anthracyclines are capable of altering iron homeostasis through the creation of Fe–anthracycline complexes and the posterior production of ROS,21 even in the absence of a reduction system (through intramolecular redox reactions) and some researchers have suggested that the toxicity of the combination of iron and some anthracyclines (doxorubicin) is not a simple additive effect.22
At the same time, iron is capable of catalyzing diverse molecular reactions that create ROS, independently of the abovementioned Fe–anthracycline complex, generating hydroxyl radicals, and it is well known that this reaction is accelerated under the influence of iron itself in the cellular environment.23
Iron regulatory proteins or IRPs (also known as iron responsive element binding proteins) are important elements in regulating iron metabolism and the interactions between doxorubicin and the human iron regulatory system have been studied by some researchers. Studies have found that the conversion of IRP1 to a null protein via doxorubicin metabolites yields a metabolic cellular impairment that may account to a certain degree for myocardial dysfunction, opening new paths towards a better understanding of the role of iron metabolism.24
Likewise, animal models have shown that iron overload is capable of potentiating doxorubicin cardiotoxicity by mechanisms other than ROS production.25 Recently, human studies have confirmed this finding, showing that even cumulative doses of doxorubicin in the “safe” range are capable of inducing higher iron levels in cardiac tissue, specially at the intracellular level, opening the possibility of cardiotoxicity related with iron deposits as a new pathophysiologic mechanism26 and raising the question of whether a real “safe” dose exists in terms of preventing long term cardiomyopathy.
Disruption of energetic mechanismsThe integrity of mitochondrial function is of cornerstone importance in the physiology of myocardial cells, as ATP production is highly dependent on this organelle (almost all of the ATP used by cardiac cells is produced by the electron transport chain).27
Several studies have suggested that disruptions in the mitochondrial membrane potential and respiratory chain are capable of inducing loss of architectural integrity, loss of energy production capability and difficulties in maintaining metabolic demands.28,29
Recently, cadiolipin, a phospholipid of utmost importance in energy metabolism and a component of the inner mitochondrial membrane, has been suggested as having a major role in anthracycline-mediated cardiomyopathy.30 Doxorubicin, by its ability to bind cardiolipin would modify membrane properties, environment and function, leading to disruption of several energetic mechanisms.
As mentioned before, no pathway by itself justifies the complete clinical picture, and the disruption of mitochondrial functions mediated by cardiolipin-doxorubicin interactions are also mediated by oxidative stress,15 in fact, the disturbance in mitochondrial permeability has been related to a direct opening of the permeability transition pore by substances formed during de redox cycling of some anthracyclines and its dysfunction could be an early indicator of doxorubicin-induced apoptosis.
Sarcomeric structure alterationsCardiotoxicity is characterized at the sarcomere level by disarray and loss of myofilaments and studies have demonstrated that anthracyclines are capable of disrupting the contractile apparatus by direct mechanisms, moving further away from the cytotoxic effects mediated by ROS.
Titin is a giant protein and integral part of the sarcomere structure, extending from the M-line to the Z-disk and has diverse functions, structural as well as dynamic and regulatory.31 The loss of integrity or function of titin has recently been shown to play an important role in the pathophysiology of dilated cardiomyopathy.32,33 Exposure to anthracyclines induces an accelerated degradation of titin through proteolytic pathways, leading to energetic compromise. In vitro studies have shown that such effect is also related with loss of structural integrity and disarray or even myocyte necrosis in a calpain-dependent way.34,35
Recently, a very interesting work by Chen et al.36 studied the role of cardiac ankyrin repeat protein (CARP or ANKRD1, a transcriptional regulatory protein that may act as a nuclear transcription factor negatively regulating the expression of cardiac genes) in the pathophysiology of anthracycline cardiomyopathy. His group found that CARP is extremely sensible to doxorubicin, leading to depletion of CARP protein levels (by inhibition of CARP transcription) and causing marked sarcomeric disarray. In their same study, doxorubicin induced cardiac transcription factor GATA4 (GATA Binding Protein 4, thought to regulate genes involved in myocardial differentiation and function, including CARP) depletion, suggesting a co-dependent role for both proteins in maintaining sarcomere structure.
Inflammatory mediatorsAnthracyclines are capable of promoting proinflammatory cytokines release, which has been related to several manifestations ranging from cardiotoxicity to asthenia experienced by these patients.37
Doxorubicin stimulates macrophages and monocytes with the subsequent histamine and tumoral necrosis factor alpha release; these substances have been related with architectural alterations and dilated cardiomyopathy through myocardial receptors binding.15
Wong et al.38 tested the effects of doxorubicin on the mitogen-activated protein kinase (MAPK) pathway, which is essential in translating signals from the cellular surface (through cellular membrane receptors) to the nucleus and, among other functions, regulates inflammatory cytokines. By using clinically relevant doses of doxorubicin, they found an increased expression of inflammatory genes in macrophages as well as increased levels of IL-1β and IL-6 in treated cells, without changes in the expression of tumoral necrosis factor (TNF). The expression of these cytokines has been shown to have a relevant role in cardiotoxicity induced by anthracyclines, mainly by modulating apoptosis through TNF receptors, whose function is affected by doxorubicin.39
Genetics and cardiotoxicityThe study and approach of several cardiovascular diseases has changed dramatically with the advent of genomic medicine, and anthracycline-induced cardiotoxicity is not alien to these advances. Previous evidence exists in terms of the usefulness of the study of genetic polymorphisms related to certain drug toxicities2,5 and some groups have investigated the possible relationship between polymorphisms (e.g. V244M on the CBR3-carbonyl reductase 3 gene) and heart failure related to these drugs.40 At the same time, certain genotypes might be associated with intracellular iron accumulation26 in patients treated with anthracycline regimens, possibly favouring cellular damage via the mechanisms above mentioned.
Wojnowski et al.41 also reported that some genetic variants in doxorubicin transport and in free radical generating enzymes might be involved in the predisposition to present doxorubicin-induced cardiotoxicity, at least in certain lymphomas.
While these observations are promising and provide new insights into different aspects of doxorubicin cardiotoxicity, with the current data, we are not able to define a clear genotype-response correlation and further investigation should try to clarify this and provide more information about the clinical usefulness of genotyping.
ConclusionsWith the improvements in early diagnosis and anti-tumoral treatments, we are witnessing an increased survival in oncologic patients. In this context, cardiologists are faced with new tasks, like how to manage the toxic effects caused by chemotherapy agents.
Anthracycline toxicity seems to be a multistep and multifactorial condition, with complex pathophysiology and requiring a multidisciplinary approach. A deep understanding of this toxicity and its mechanisms will possibly allow us to reduce its incidence, identify early markers of susceptibility or find more specific therapeutic targets. As we have reviewed in the present paper, no mechanism seems capable by itself of causing the whole clinical picture.
Development of new anticancer therapies will surely bring further challenges to cardiologists in the next decade, as the advent of novel therapies in oncology will also change the current landscape of these toxicities. It seems obvious to us that the “specialty” of cardiooncology is much needed in the current context.
FundingThere was no funding for this research.
Conflict of interestAuthors do not have any conflict of interests regarding this work.