


Dentro de las malformaciones vasculares de las arterias en el tórax encontramos: 1) doble arco aórtico; 2) arco aórtico derecho y conducto arterioso izquierdo persistente; 3) arteria subclavia derecha aberrante; 4) arteria pulmonar izquierda aberrante, y 5) arteria innominada anómala. Se revisan los pacientes con arteria subclavia derecha aberrante y su manejo.
MétodosSe estudiaron en forma retrospectiva los expedientes de 29 pacientes en edad pediátrica, de enero de 1992 a diciembre 2012, con las siguientes variables: edad de inicio de los síntomas, manifestaciones clínicas, defectos cardiovasculares, método diagnóstico y abordaje quirúrgico.
ResultadosEl mayor número de pacientes cursó de forma asintomática, únicamente el 31% lo hizo con síntomas durante el primer año de vida, y se llevó a cabo el diagnóstico en un 35% mediante cateterismo. Fue la persistencia del conducto arterioso la cardiopatía más frecuente, con un 13%; el síndrome de Down se encontró en un 21% de los pacientes. El tratamiento más utilizado fue la sección de la arteria subclavia aberrante para la liberación del esófago.
ConclusionesEs importante la sospecha diagnóstica en pacientes con sintomatología durante la alimentación, con trastornos de la deglución con sólidos y, en algunos casos, con disfagia o hasta con dificultad respiratoria. Un número significativo de estos pacientes no son diagnosticados oportunamente, algunos alcanzan la edad adulta sin diagnóstico. El hallazgo de esta malformación ocurre durante los estudios de imagen, cuando se evalúa la aorta o en el estudio del reflujo gastroesofágico, ya que con el trago de bario se aprecia la compresión extrínseca del esófago.
Congenital vascular malformations of the major arteries in the chest have been classified into 5 groups: 1) double aortic arch; 2) right aortic arch with left ligament or persistent ductus arteriosus; 3) aberrant subclavian artery; 4) aberrant left pulmonary artery, and 5) anomalous innominate artery. We reviewed the patients with aberrant right subclavian artery and their treatment.
MethodsWe studied retrospectively the records of 29 patients with aberrant right subclavian artery in childhood, from January 1992 to December 2012, analyzing the following variables: age at onset, clinical manifestations, associated cardiovascular defects, diagnosis and surgical approach method.
ResultsWe found that most patients have an asymptomatic course, only 31% of them course with symptoms during the first year of life, with an incidental diagnosis of 35% during catheterization or other imaging studies. Patent ductus arteriosus was the most frequently associated congenital malformation, with 13%. Down's syndrome was found in 21%. The most common treatment was surgical section of the aberrant subclavian artery to release the esophagus.
ConclusionsThis vascular abnormality must be suspected in those patients with dysphagia, dyspnea, chest pain during feeding or breathing difficulties. A significant number of patients are not diagnosed in time, some reach adulthood without a diagnosis. This malformation is often found in imaging studies when evaluating the aorta or in a gastroesophageal reflux study, in which the barium bolus reveals the extrinsic compression of the esophagus.
Las malformaciones vasculares de las arterias mayores en el tórax han sido clasificadas en 5 grupos principales: 1) doble arco aórtico; 2) arco aórtico derecho con un ligamento o conducto arterioso izquierdo persistente; 3) arteria subclavia aberrante; 4) arteria pulmonar izquierda aberrante, y 5) arteria innominada anómala.
Algunas anomalías vasculares que involucran el arco aórtico no forman propiamente un anillo vascular; sin embargo, generan compresión de la vía aérea o el esófago, por lo que han sido agrupadas descriptivamente dentro del grupo de los anillos vasculares, clasificándose de diversas formas, dependiendo de la posición del arco aórtico con respecto a la tráquea1.
Los 2 tipos más comunes de anillos vasculares son el doble arco aórtico y el arco aórtico derecho con arteria subclavia izquierda aberrante y ligamento arterioso izquierdo; constituyen entre el 85-95% de los casos. Los otros 2 tipos de anillos vasculares, extremadamente raros, son el arco aórtico derecho con disposición en espejo de los vasos supraaórticos con respecto al arco aórtico izquierdo y ligamento arterioso retroesofágico, y el arco aórtico izquierdo con aorta descendente y conducto arterioso derechos y arteria subclavia derecha retroesofágica. La arteria pulmonar izquierda anómala o sling de la arteria pulmonar representa el 10% de los casos, se origina de una anormalidad del sexto arco branquial y produce un anillo completo. Otras anomalías que producen síntomas, pero no forman anillo vascular completo, incluyen la arteria subclavia derecha retroesofágica con arco aórtico a la izquierda y la arteria innominada anómala2–6.
Estas anomalías en el desarrollo del arco aórtico suceden en el 1 a 2% de la población, habitualmente como variantes de la normalidad. Sin embargo, una arteria subclavia derecha aberrante ha sido reconocida y encontrada en al menos el 0.4 de las autopsias estudiadas2. En los fetos sin anomalías cromosómicas ni cardiacas, la frecuencia de arteria subclavia derecha aberrante es de 0.57.
Las anomalías del arco aórtico y sus grandes ramas ocurren en aproximadamente un 1% de los pacientes con enfermedad cardiaca congénita. Grandes institutos de referencia informan entre 1 y 8 (media de 2) de estos pacientes por año8.
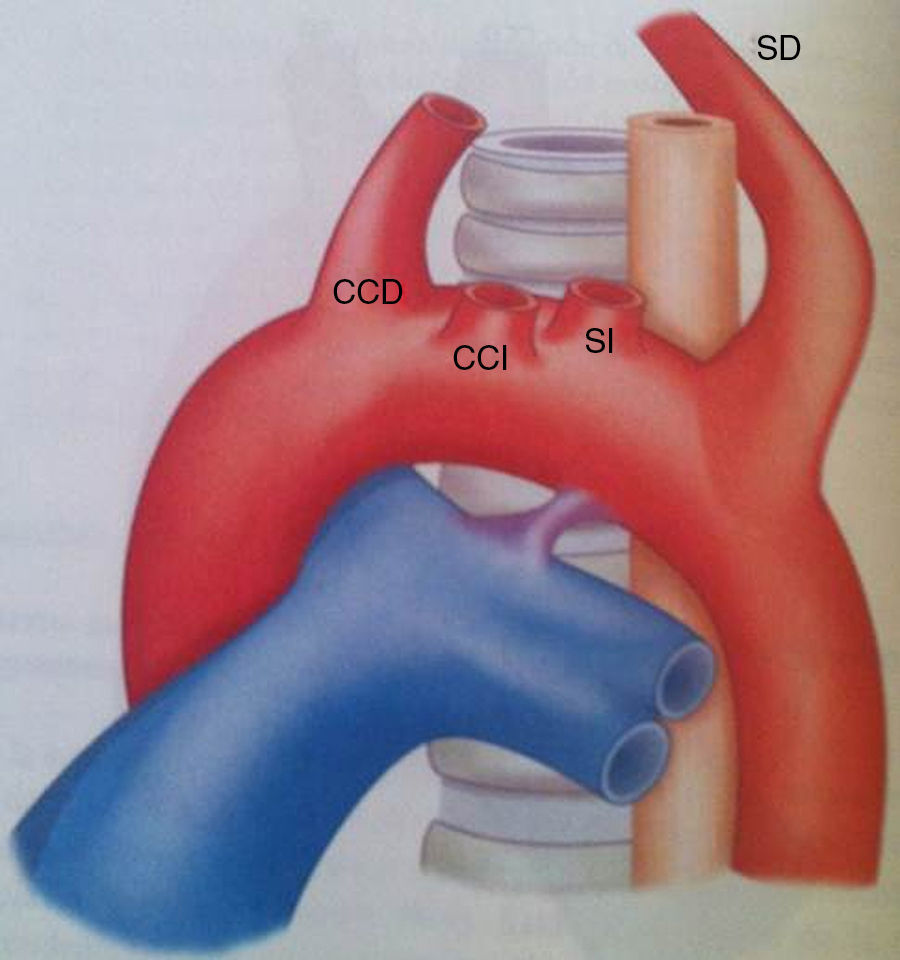
Entre las alteraciones del patrón de ramificación del arco aórtico fetal se encuentra el arco aórtico izquierdo con arteria subclavia derecha aberrante (fig. 1). Esta anomalía resulta de la regresión temprana del cuarto arco aórtico derecho, entre la carótida común y la subclavia derecha. Si el ligamento arterioso derecho involuciona, no se forma un anillo vascular, pero si este persiste, su curso puede ser desde la subclavia aberrante a la arteria pulmonar derecha y forma un anillo completo.

Embriológicamente, en pacientes con arco aórtico izquierdo, una arteria subclavia aberrante resulta de la obliteración del cuarto arco faríngeo derecho. Por tanto, la arteria subclavia derecha pierde su conexión con la arteria innominada y nace de la aorta descendente, a menudo de un remanente de la aorta dorsal derecha persistente, llamado divertículo de Kommerell. En el arco aórtico interrumpido se ha demostrado que la obliteración del cuarto arco faríngeo izquierdo resulta en una interrupción tipo B (el segmento entre la subclavia izquierda y la arteria carótida común izquierda en el arco izquierdo)9. Esta tendencia a la involución bilateral de los cuartos arcos explica la alta incidencia de arteria subclavia aberrante en pacientes con esta cardiopatía.
Pese a los amplios límites de incidencia de la arteria subclavia derecha aberrante, de acuerdo con lo publicado, es posible que en la población general su incidencia esté subestimada, ya que suele ser asintomática y ello impide su consideración clínica.
Los pacientes que presentan esta anormalidad vascular pueden mostrar compresión traqueoesofágica, principalmente esofágica. Dentro de los síntomas y signos se incluyen estridor, dificultad respiratoria, disfagia e infecciones respiratorias recurrentes por reflujo. Pacientes con anillo vascular completo usualmente son sintomáticos desde el nacimiento.
El diagnóstico de arteria subclavia derecha aberrante se basa en la metodología descrita por Chaoui et al.9. Tras obtener un corte axial del tórax fetal, a nivel del plano de 3 vasos-tráquea, se logra visualizar la salida de un tronco vascular desde el arco aórtico, próxima a la unión de este con el ductus arterioso, y con un trayecto que, de izquierda a derecha, discurra posterior a la tráquea en dirección al hombro derecho fetal. En la última década, la importancia del hallazgo ecográfico prenatal de la arteria subclavia derecha aberrante reside en su asociación con cromosomopatías o cardiopatías congénitas, especialmente con la trisomía 21 y la microdeleción 22q11 (síndrome de DiGeorge), y otros síndromes genéticos asociados con anomalías del arco aórtico10–12.
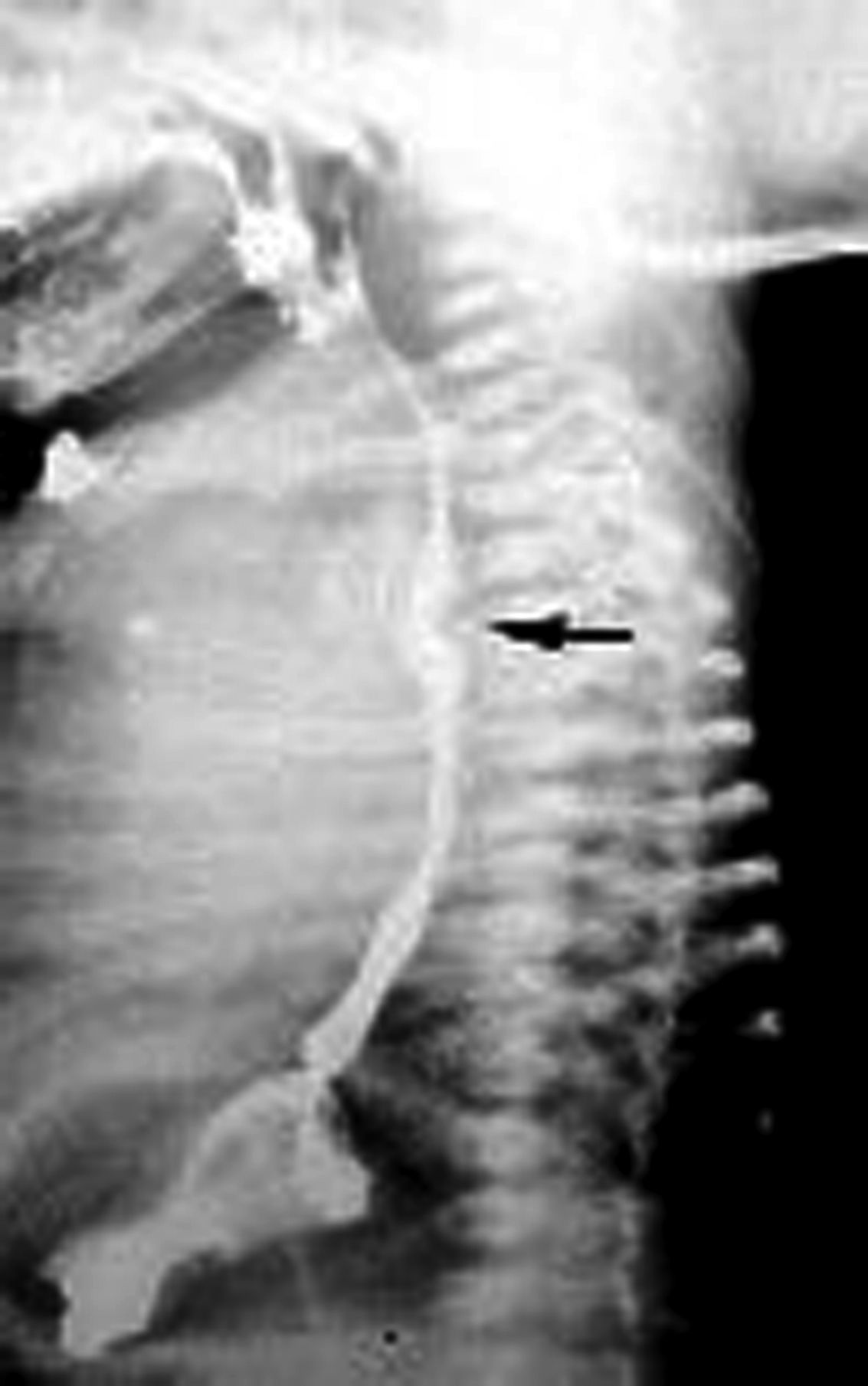
El estudio o abordaje de esta alteración en el periodo posnatal se inicia con la realización de un esofagograma con bario, en donde se observa la presencia de una muesca posterior de forma cóncava en la porción media del esófago (fig. 2).
, producto de la compresión extrínseca por arteria subclavia derecha aberrante.")
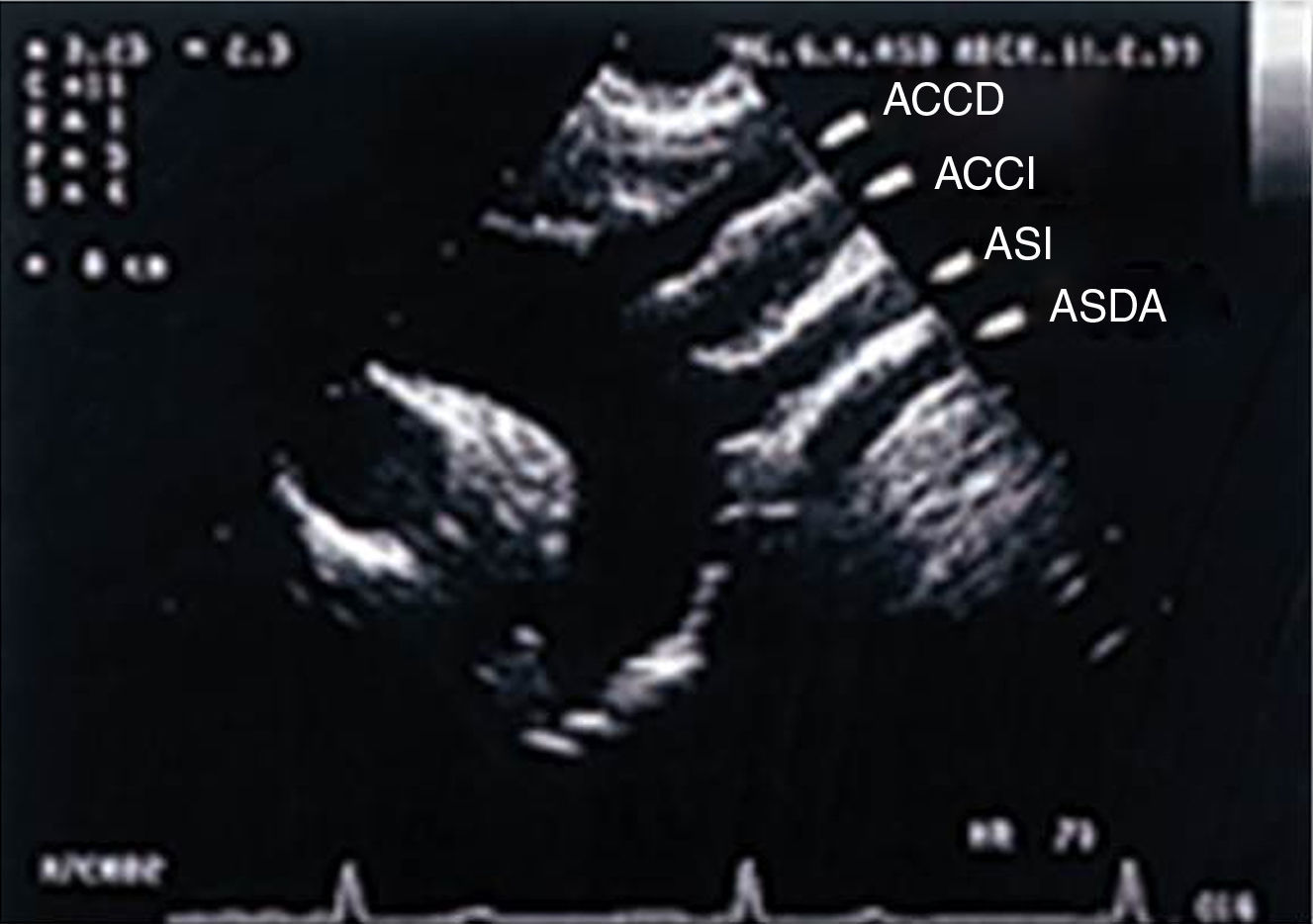
El ecocardiograma bidimensional, en la proyección supraesternal en un corte longitudinal del arco, permite observar un arco aórtico izquierdo del que nacen 4 troncos supraaórticos (fig. 3).
.")
La resonancia magnética ha sido un estudio de imagen de gran utilidad para evaluar a estos pacientes. Sin embargo, con el reciente incremento en la velocidad de adquisición y, además, la reducción en la necesidad de sedación para los pacientes estudiados, la tomografía helicoidal juega un rol importante en la evaluación de estas personas13–15. Aunque no se ha comprobado la asociación entre la arteria aberrante prenatal aislada y la microdeleción del cromosoma 22q11, su detección en cardiopatías conotruncales es el marcador más importante de esta alteración genética, por lo que se recomienda su estudio. Informes recientes enfatizan la alta prevalencia de arteria subclavia aberrante en pacientes con deleción del cromosoma 22q1113–15.
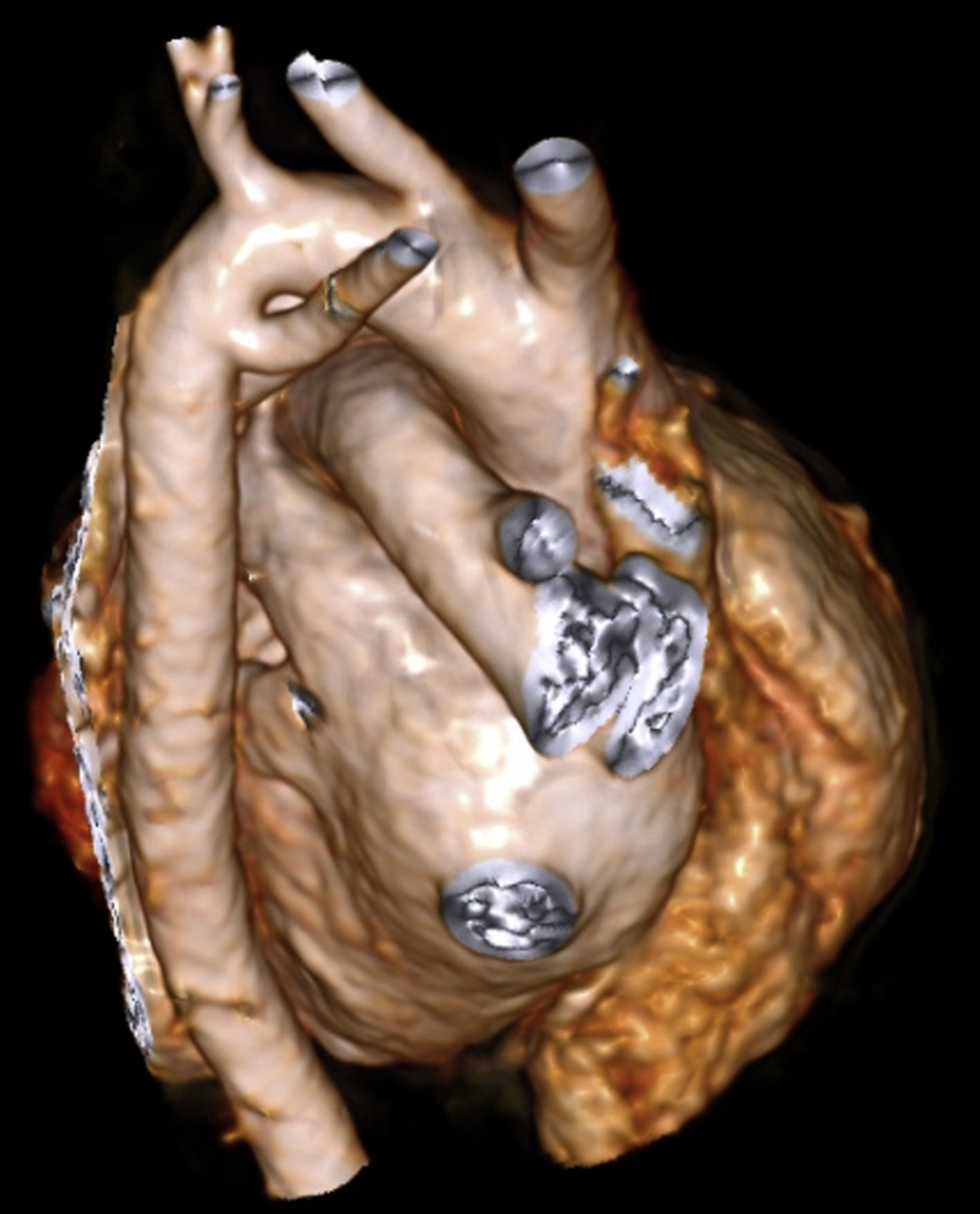
En una reconstrucción volumétrica se observaba el nacimiento independiente de los 4 troncos, siendo la arteria subclavia derecha aberrante el último vaso que emerge del arco. Este vaso aberrante nace del segmento posteroinferior del arco aórtico distal y cruza hacia el lado derecho posterior a la tráquea (fig. 4).
. Se observan 4 troncos supraaórticos independientes naciendo del cayado, siendo el último vaso la arteria subclavia derecha aberrante.")
La asociación más conocida con arteria subclavia aberrante es la tetralogía de Fallot con una arteria subclavia derecha aberrante, y fue comentada por Blalock16 en 1948. Otros autores estudiaron la asociación en sus series17–21. La arteria subclavia aberrante es rara en pacientes con transposición clásica de las grandes arterias y doble vía de salida del ventrículo derecho sin obstrucción izquierda.
Dentro de los diagnósticos diferenciales de esta enfermedad debemos tomar en cuenta que la muesca en la cara posterior del esófago limita las posibilidades de origen vascular.
La cirugía correctiva se encuentra indicada en todos aquellos pacientes que presentan sintomatología obstructiva aérea o durante la deglución, así, también se considera indicación absoluta en los casos con arteria subclavia derecha aberrante aneurismática.
Muchas técnicas quirúrgicas, como la esternotomía media, las toracotomías izquierda y derecha, así como la incisión supraclavicular, han sido utilizadas para cumplir este objetivo.
Gross, en 1946, realizó la primera cirugía correctiva para esta anomalía. La técnica quirúrgica en pacientes pediátricos es la siguiente: se inicia el abordaje a través de una toracotomía anterolateral derecha en el cuarto espacio intercostal. Los nervios vago y laríngeo recurrente son identificados y preservados. El vaso anómalo se identifica y se diseca desde su posición retroesofágica y, posteriormente, se secciona de su origen en el arco aórtico. Después de su sección, el cabo proximal se retrae hacia la región posterior del esófago22,23.
La arteria subclavia puede ser ligada y seccionada impunemente sin que se comprometa la circulación al miembro torácico que irriga, ya que en la mayoría de los casos, la circulación colateral proveniente del sistema vertebrobasilar es suficiente para mantener la viabilidad y el desarrollo24.
MétodosEl estudio que se realizó es una investigación de tipo retrospectivo, descriptivo. Se revisó la base de datos del Instituto Nacional de Cardiología de forma retrospectiva, analizando en total 900 expedientes, de los cuales se obtuvieron 29 pacientes con diagnóstico de arteria subclavia derecha aberrante que cumplían con los criterios de inclusión establecidos, en el periodo de enero de 1992 a diciembre de 2012.
Incluimos a pacientes en edad pediátrica (desde neonatos hasta 18 años de vida), los cuales se dividieron en 2 grupos: en el primero, los menores de un año de edad; en el segundo, pacientes mayores de un año de edad en el momento del diagnóstico. Los criterios de exclusión fueron: pacientes portadores de arteria subclavia izquierda aberrante.
A la población en estudio se le analizaron las siguientes variables de forma descriptiva: frecuencia, distribución, sintomatología, defectos asociados, método diagnóstico y abordaje quirúrgico.
En cuanto al análisis estadístico, las características clínicas y demográficas fueron expresadas como frecuencias y porcentajes para variables categóricas, y media y desviación estándar para variables continuas distribuidas de forma gaussiana. En las variables de distribución no paramétrica se utilizaron medianas y rangos, así como pruebas de análisis de medias, con χ2 y prueba exacta de Fisher. El análisis de sobrevida fue realizado en relación con el género y la intervención quirúrgica para un tiempo de seguimiento de 250 meses (20 años) por el método de Kaplan y Meier. El software utilizado fue SPSS® versión 19.
ResultadosDe los pacientes analizados, 10 (34%) eran niños, y 19 (66%), niñas. La edad en años y meses fue de una mediana de 6 con rango de 0.9 meses a 11 años; 9 pacientes (31%) del total iniciaron sintomatología en el primer año de vida, la cual se presentó con mayor frecuencia como disfagia en 5 de ellos y estridor en 4. Ningún paciente inició sintomatología después del primer año de vida y 20 (69%) cursaron asintomáticos; el diagnóstico se hizo en un 35% por medio de cateterismo como principal herramienta diagnóstica, siguiendo en frecuencia la tomografía (20%) como método diagnóstico de esta enfermedad.
Las anomalías congénitas cardiovasculares asociadas se documentaron en 28 pacientes (96.5%), siendo en orden de frecuencia las siguientes:
- -
Persistencia de conducto arterioso en 4 pacientes (13.7%).
- -
Comunicación interventricular asociada a comunicación interauricular en 3 pacientes (10.3%).
- -
Tronco arterioso común en 3 pacientes (10.3%).
- -
Coartación aórtica asociada a estenosis aórtica y conducto arterioso persistente en 3 pacientes (10.3%).
- -
Defecto de la tabicación auriculoventricular en 2 pacientes (6.9%).
- -
Tetralogía de Fallot en 2 pacientes (6.9%).
- -
Interrupción del arco aórtico en 2 pacientes (6.9%).
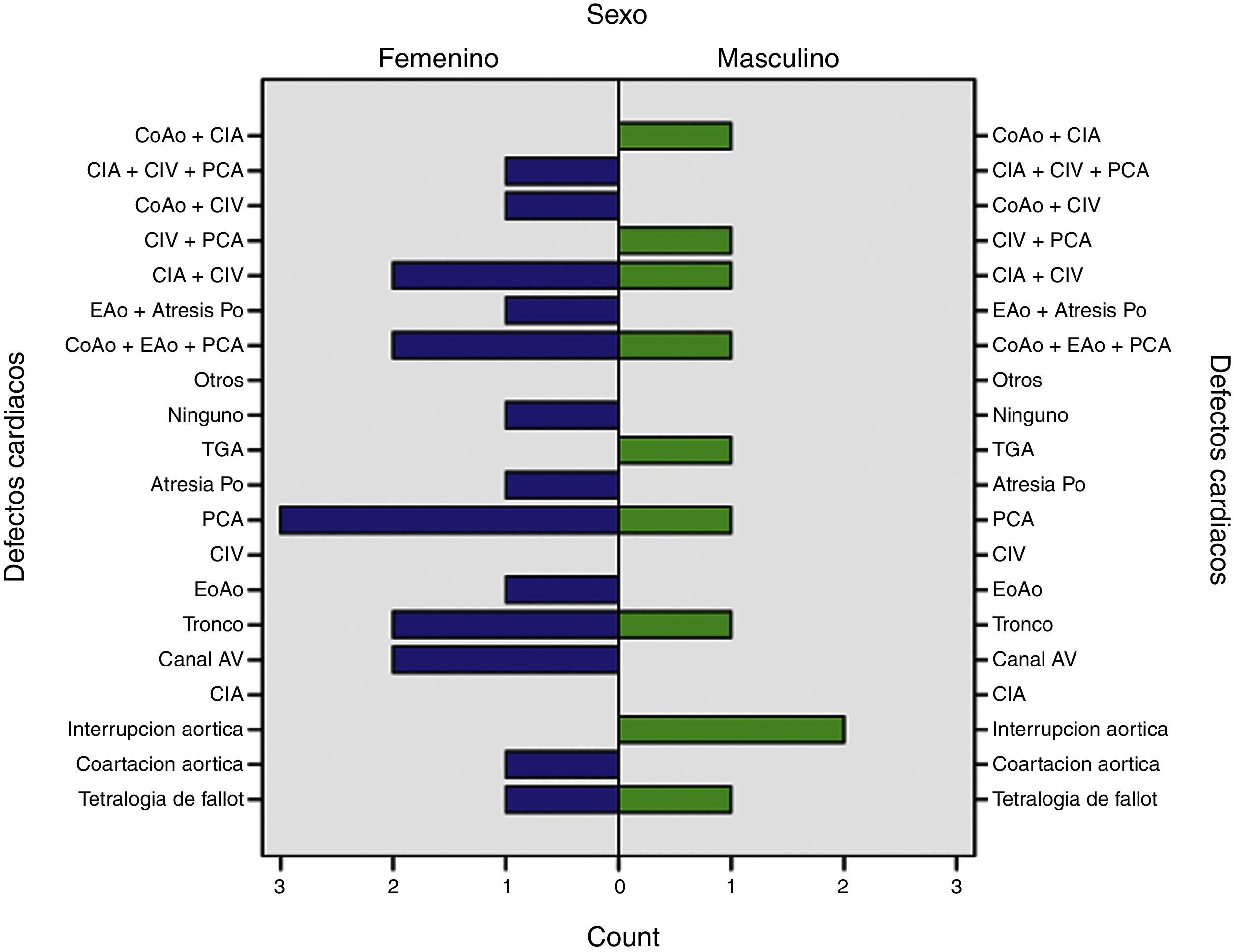
Continuaron en frecuencia la atresia pulmonar con septum interventricular intacto; coartación aórtica; estenosis aórtica; transposición de grandes arterias; y asociaciones como coartación de aorta con comunicación interauricular; conducto arterioso persistente con comunicación interventricular e interauricular; coartación de aorta con comunicación interventricular; conducto arterioso persistente con comunicación interventricular; atresia pulmonar con comunicación interventricular y estenosis aórtica, todos ellos en un paciente cada uno (3.5%). Solo en un paciente no se documentó la presencia de cardiopatía congénita asociada (fig. 5).

Los síndromes asociados fueron: trisomía 21 en 6 pacientes (21%), síndrome de deleción del cromosoma 22q11.2, y el síndrome de Marfan, encontrados en un paciente cada uno (3%). Además, se encontró una asociación con arco aórtico a la derecha en un solo paciente de los 29 estudiados. Será necesario revisar y evaluar a pacientes con arco aórtico derecho, interrupción del arco, tronco común y el espectro amplio de la tetralogía de Fallot, pues algunos de ellos pueden formar parte del síndrome de deleción 22q11.2, y esto será motivo de otra comunicación.
El abordaje quirúrgico fue llevado a cabo en 15 pacientes, lo que representó el 52% del total, siendo manejados de forma conservadora 14 (48%). En los primeros, la técnica utilizada fue la sección de la arteria subclavia derecha aberrante con la finalidad de liberar el esófago.
Fallecieron 3 pacientes, lo cual representó casi el 10% del total de ellos; el defecto menos significativo era la arteria anómala, tenían transposición de las grandes arterias, atresia pulmonar sin comunicación interventricular, y uno de ellos, síndrome de Down y defecto de la tabicación auriculoventricular.
El promedio de tiempo de evolución y seguimiento del padecimiento con subclavia aberrante, entre los fallecidos y los que sobreviven, mostró una mediana en meses de 3 (rango 1-12) y de 14 (rango 1-245), respectivamente.
Actualmente se encuentran bajo seguimiento por la consulta externa 26 pacientes, que se encuentran en clase funcional i y ii (NYHA y modificada por Ross), sin síntomas de dificultad para deglución o respiratoria.
DiscusiónLas malformaciones del arco aórtico y de sus grandes ramas ocurren en el 0.5% de la población. La arteria subclavia derecha se origina de la aorta descendente, y sigue un curso retroesofágico, pudiendo sufrir compresión en grados variables. Es una forma de anillo incompleto, la mayoría de los pacientes se encuentran asintomáticos, pero pueden tener disfagia y no es infrecuente que no se diagnostique hasta la edad adulta1.
En este estudio se encontró que el 69% de los pacientes cursaron de forma asintomática y 9 (31%) iniciaron con disfagia durante el primer año de vida. Roofthooft et al.25 comentaron que únicamente el 20% de los pacientes con diagnóstico de arteria subclavia derecha aberrante cursaban con dificultad en la alimentación.
En la presente investigación, se encontró que el estudio más utilizado para el diagnóstico de arteria subclavia derecha aberrante fue en un 35% el cateterismo cardiaco realizado para estudio de la participación cardiaca en otras malformaciones cardiovasculares, y también la tomografía computarizada como método diagnóstico.
La malformación más asociada con la arteria subclavia derecha aberrante fue el síndrome de Down, en un 21% (6 pacientes), lo que concuerda con estudios previos de Chaoui et al. y los más recientes de Borenstein y Minekawa26, en donde se ha establecido una asociación de arteria subclavia derecha aberrante con trisomía 21 del 30%. La deleción del cromosoma 22q11 y el síndrome de Marfan se encontraron en un 3% de todos los pacientes. En informes recientes se ha enfatizado la alta prevalencia de arteria subclavia aberrante en pacientes con deleción del cromosoma 22q11.2, lo que ha determinado una nueva valoración integral de nuestros pacientes, en los cuales hay datos para sospechar la deleción, como serían los casos de interrupción del arco aórtico, el tronco común, las variedades de tetralogía de Fallot y el arco aórtico derecho. El síndrome de Marfan coincide en el 3% de nuestros pacientes con esta asociación.
Encontramos la asociación con la persistencia del conducto arterioso en un 13.7%. La cardiopatía asociada más conocida con arteria subclavia aberrante es la tetralogía de Fallot, que fue comentada por Blalock16 en 1948; Pattinson17 y Pattinson y Emanuel18 informaron una incidencia del 16% basada en series de 43 casos y en autopsias; Zapata et al.19 comentaron una incidencia del 5% en 355 especímenes con esta asociación. Más recientemente, Nakajima et al.20 informaron una prevalencia del 5%, y de más del 16% para la tetralogía de Fallot con atresia pulmonar.
La prevalencia de arteria subclavia aberrante en pacientes con tetralogía de Fallot en el estudio publicado por Ramaswamy et al.21 fue del 8%. Encontramos esta asociación en 2 pacientes (6.9%), que difiere de lo encontrado en series previas. Igualmente, las lesiones obstructivas izquierdas aisladas (coartación de aorta, estenosis aórtica e interrupción de arco aórtico) fueron encontradas en 3 pacientes (10.3%) del total del universo, siendo la tercera cardiopatía asociada a la arteria subclavia derecha aberrante, lo que concuerda con el estudio de Ramaswamy et al.21, en donde las lesiones obstructivas izquierdas aisladas fueron, de las lesiones intracardiacas, las segundas más frecuentes en pacientes con arco aórtico izquierdo y arteria subclavia derecha aberrante. Sin embargo, Zapata et al.19 encontraron que las lesiones obstructivas a este nivel se daban únicamente en un 2% de los pacientes con estas características.
La arteria subclavia aberrante es una anomalía congénita rara que usualmente no produce sintomatología. Sin embargo, los pacientes sintomáticos requieren intervención quirúrgica. En nuestro estudio, a 15 pacientes (52%) se les realizó tratamiento quirúrgico, manejándose de forma conservadora los demás; la técnica más utilizada fue la sección de la arteria subclavia derecha aberrante, con un 67% (10 pacientes), y en segundo lugar, la ligadura de la misma en 5 pacientes (33%). En el estudio realizado por Atay et al.23, los pacientes fueron tratados de forma exitosa con la sección de la arteria subclavia derecha aberrante y la reimplantación de la misma a la arteria carótida común derecha o a la aorta descendente, sin morbilidad o mortalidad quirúrgica tardía. Así, también, en la publicación de series de casos más grande que existe, Kieffer et al.24 incluyeron 33 casos con diferentes estrategias terapéuticas, informando como la técnica más utilizada la sección de la arteria subclavia derecha aberrante. Esto concuerda con nuestro estudio, apoyando lo informado en la literatura ya publicada.
ConclusionesEn el Instituto Nacional de Cardiología Ignacio Chávez la mayoría de nuestros pacientes tuvieron un curso clínico asintomático en los últimos 20 años. Se han estudiado 29 pacientes con esta enfermedad; la serie más grande evaluada ha sido de 33 pacientes, por Kieffer et al.24. La toma de decisiones asertivas en cuanto al plan que se debe tomar con estos pacientes, así como el discernir entre otorgar un tratamiento conservador o bien uno correctivo, que en nuestra serie fue la sección de la subclavia para liberar el esófago y evitar problemas isquémicos en el brazo derecho por esta conducta, debe ser tomado en cuenta el riesgo y beneficio que ofrece cada uno de ellos.
Al conocerse la asociación de esta anomalía con otras malformaciones congénitas cardiovasculares y síndromes, como la trisomía 21 o la deleción del cromosoma 22q11.2, aumentan las posibilidades de un diagnóstico temprano. Por tanto, el hallazgo de arteria subclavia derecha aberrante debe alertar sobre la posibilidad de realizar un estudio genómico dentro del abordaje integral de estos pacientes. Así también, es importante tener sospecha diagnóstica de este padecimiento cuando los pacientes presentan trastornos de la deglución o cursan con dificultad respiratoria, ya que un número significativo de pacientes que llegan a la edad adulta no fueron diagnosticados.
FinanciaciónNo se recibió patrocinio de ningún tipo para llevar a cabo este estudio/artículo.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.