


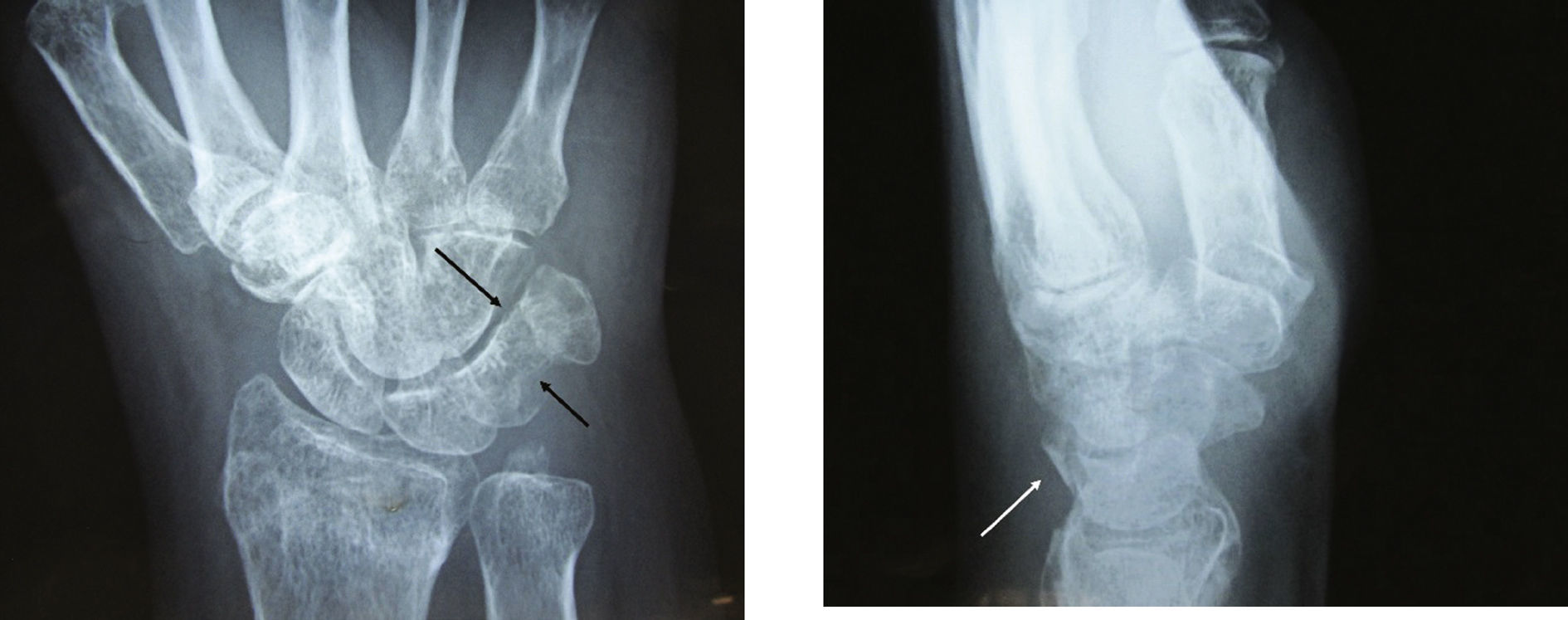
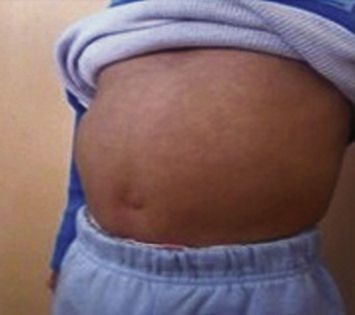
Paciente femenino de 40 años de edad, sufrió caída sobre mano derecha en dorsi-flexión y desviación cubital.
Imagen Aten Fam. 2016;23(3):116.
Respuesta del diagnóstico
Prune Belly
La imagen correspondió a un paciente con diagnóstico de síndrome de Prune Belly, en él se apreció la ausencia de músculos abdominales al palparse directamente asas intestinales observándose borde hepático y red venosa colateral; además cabalgamiento de cuarto ortejo sobre tercero de pie izquierdo.
El paciente acudió a la consulta por múltiples infecciones urinarias secundarias a reflujo vesiculoureteral, abdomen distendido y criptorquidia bilateral, la cual le fue reparada a los cuatro años.
El síndrome de Prune Belly conocido también como síndrome de Eagle-Barret, síndrome de la triada, síndrome de displasia mesenquial o “vientre en ciruela pasa” en su traducción, fue descrito por primera vez en 1839 por Frolich quien registró el caso de un neonato masculino con agenesia de la musculatura abdominal predominantemente de músculos abdominales inferiores (rectos inferiores y oblicuos) y criptorquidia bilateral, añadiéndose en 1895 por Parker, malformaciones del tracto urinario tales como: dilatación megaloquística de uretra, vejiga y uréteres, reflujo vesiculoureteral, obstrucción e hidronefrosis, completando la triada clásica de este síndrome (hipoplasia o agenesia de musculatura abdominal, criptorquidia bilateral y alteraciones en vía urinaria).1,2
Se estima que actualmente existen un poco más de 300 casos reportados en la literatura médica, su incidencia es de uno por cada 40 000 recién nacidos vivos, siendo más frecuente en hombres que en mujeres con una proporción de 18:1, solo reportándose de 3 a 5% en mujeres (pseudo-pseudo Prune Belly), 20% fallece antes del primer mes y 50% antes de los dos años, el restante 30% normalmente no alcanzará la adolescencia, pues compromete su vida el daño renal y la displasia pulmonar, principalmente.3
La etiología actualmente se desconoce, pero se ha asociado a alteraciones genéticas por daño en mesodermo, infecciones y factores mecánicos diversos como edema intraparto. Actualmente existen ensayos que sugieren la existencia de alguna forma de transmisión hereditaria, principalmente la recesiva ligada al cromosoma X, aunque otros patrones como el autosómico recesivo, dominante o mitocondrial también han sido propuestos. En el desarrollo de este síndrome se ha observado la prevalencia de la mutación del gen chrm3 del cromosoma 1q43. Se menciona también, que tras las alteraciones renales como megaureteres se genera un efecto masa en el abdomen con hipoplasia de músculos abdominales, lo cual provoca la dilatación abdominal característica en este síndrome.4
El diagnóstico se realiza con una ecografía prenatal de la semana 18 a la 22, en la que se observa el aumento del tamaño de vejiga y uréteres e hidramnios, el diagnóstico de certeza se realizará con el recién nacido, observando el abdomen distendido, con piel arrugada y ausencia de testículos en bolsas escrotales.1,5
El síndrome de Prune Belly se clasifica en: síndrome Prune Belly clásico (triada clásica), Pseudo Prune Belly (ausencia de criptorquidia), Pseudo-Pseudo Prune Belly (ausencia de criptorquidia y ausencia de alteraciones renales o alteraciones renales leves -visto en mujeres-) y la variante letal del síndrome de Prune Belly, la cual es aquella con obstrucción anatómica uretral con importantes repercusiones en vejiga, uraco, uréteres y riñón condicionando a una muerte neonatal.6
De acuerdo con la valoración de la función renal, se divide en tres categorías: 1. Buena, con valores de uremia o creatinina normales; 2. Intermedia, con uremia elevada; y 3. Pobre, con uremia francamente alta.7,8
Los signos y síntomas encontrados en estos pacientes son los relacionados con su triada clásica añadiéndose la existencia en algunos casos de hipoplasia pulmonar, retraso mental o capacidades intelectuales intactas, ascitis, anomalías de extremidades, displasia de caderas, ano imperforado, polidactilia, sindactilia, anomalías gastrointestinales como mal rotaciones, atresias y bandas intestinales, paladar hendido, hernia de Bochdaleck y alteraciones bucales.7,9,10
El tratamiento a corto y mediano plazo inminentemente es quirúrgico, pero se debe evaluar en primera instancia la función cardiorrespiratoria y función renal, valorando la uremia o creatinina, electrolitos y urocultivo, así como procesos infecciosos; para posteriormente determinar qué cirugía se efectuará primero. Cabe señalar que al nacer no se realiza cirugía de urgencia, ya que podría agravar el caso y empeorar el pronóstico si se hace precozmente. Se puede efectuar reconstrucción abdominal, orquidopexia y reconstrucción pielo-uréter-vesico-uretral, según sea el caso.1,9,10
El tratamiento general, sin duda, debe ser multidisciplinario con la finalidad de brindar una mejor calidad de vida, tanto para el paciente en primera instancia, como para su familia.
Sugerencia de citación: Ávila-Martínez JC, Delgado-Quiñones EG, Uriostegui-Espíritu LC. Síndrome de Prune Belly. Aten Fam. 2016;23(4):162-163.