







Hoy día nadie discute que los datos de eficacia (resultados obtenidos con un medicamento en condiciones ideales, controladas y experimentales) deben complementarse con los datos de efectividad (resultados obtenidos con un medicamento en condiciones reales de uso, en la práctica médica habitual), si se quiere conocer los efectos terapéuticos reales y finales de los medicamentos en las enfermedades a tratar y en la salud y el bienestar de los pacientes1.
Por lo tanto, es necesario conocer cuál es el efecto de los medicamentos cuando se utilizan en el mundo real (donde hay pacientes polimedicados y con múltiples enfermedades asociadas, así como niños, ancianos y embarazadas) y cuando se emplean en condiciones en las que el incumplimiento terapéutico es elevado y en regímenes terapéuticos crónicos, donde los pacientes tomarán el medicamento durante largos períodos2.

Los estudios de efectividad (también denominados naturalísticos o naturalistas) no tienen un diseño estándar y se pueden emplear diseños prospectivos, bien estudios observacionales (básicamente, estudios de cohortes) o bien estudios aleatorizados (ensayos clínicos pragmáticos) y, además, se pueden usar también diseños retrospectivos, realizados sobre todo a través de las bases de datos con historias clínicas informatizadas3.
Aunque los ensayos clínicos pragmáticos (que reflejan la práctica médica habitual) presentan la mayor validez interna (al aleatorizar a los pacientes que entran en el estudio a uno u otro tratamiento) y, por tanto, a priori deberían ser los diseños de elección a la hora de medir la efectividad de los medicamentos, en muchas ocasiones es necesario recurrir a los estudios observacionales para su evaluación, como en los casos en los que efectuar ensayos clínicos sea poco ético, difícil de ejecutar o suponga un importante retraso para el conocimiento de los resultados4.
Los estudios observacionales presentan una alta validez externa y son generalizables a toda la población, ya que en ellos se incluye a todo tipo de pacientes y se evalúa la efectividad en la práctica clínica real5, aunque siempre habrá una mayor probabilidad de que los grupos que se comparen no sean totalmente homogéneos en cuanto a sus características sociodemográficas, las comorbilidades asociadas y los factores pronósticos. Por ello, siempre será necesario efectuarlos con una altísima rigurosidad científica y con el empleo de técnicas que nos permitan incrementar la homogeneidad de los grupos en estudio. Si estos estudios se realizan siguiendo estos altos estándares de calidad, se ha comprobado que los resultados son bastante similares a los que se pueden extraer de un ensayo clínico y, desde luego, complementarios6.
Dada la creciente demanda de datos de efectividad por parte de los diferentes agentes decisores, el objetivo de este trabajo se centra en repasar los pasos que se deberían seguir a la hora de diseñar estudios observacionales y en crear unas listas-guía que actúen como recomendaciones a la hora de su elaboración, realización y análisis, con el fin de que se incremente la validez y precisión de sus resultados, así como su credibilidad y relevancia como datos de apoyo en la toma de decisiones en política farmacéutica.
Principios y fundamentos a la hora de diseñar y/o valorar un estudio observacional para evaluar la efectividad
Aunque hay distintos tipos de estudios observacionales, en este artículo solamente se revisarán los estudios de cohortes, ya que son los que más se emplean para valorar y cuantificar la efectividad. El resto de los posibles diseños, como los estudios de casos y controles y los estudios transversales, rara vez utilizan para medir efectividad (aunque en alguna circunstancia puedan usarse para este propósito) y se emplean fundamentalmente para evaluar efectos adversos (estudios de casos y controles) y la prevalencia de diferentes estados de salud (estudios de prevalencia).
Con el fin de facilitar la aplicación de unos criterios metodológicos correctos, se han elaborado unas listas-guía de recomendaciones para poner en marcha un estudio de efectividad en las que se distinguen los diseños prospectivos y retrospectivos (tablas 1 y 2).

Estudios de cohortes prospectivos
En estos estudios se selecciona a una cohorte de pacientes sobre la base de que esté recibiendo el medicamento que se quiere evaluar, y a otra (cohorte control) que esté tomando otro u otros medicamentos (fármacos comparadores), lo que permitirá comparar ambas cohortes en cuanto al grado de efectividad alcanzado. Las ventajas más importantes de los diseños prospectivos son: a) se pueden definir de antemano las características de los pacientes que serán incluidos en las cohortes; b) permiten recoger información de los pacientes de gran interés (medicación concomitantes, gravedad de la enfermedad, grado de bebedor y fumador, comorbilidades asociadas, etc.); c) permiten fijar cómo se va a evaluar la efectividad (qué variables van a ser valoradas), y d) se puede determinar, a priori, cómo y en qué momento recogerá la información de ambas cohortes durante el estudio.
Cuando se vaya a elaborar y efectuar un diseño de cohortes prospectivo, siempre será deseable seguir las siguientes pautas y recomendaciones:
1. Definir el objetivo y la razón de ser del estudio, así como las variables que se van a medir para conocer la efectividad. Será necesario especificar las razones por las que se va a realizar el estudio y definir con claridad sus objetivos y finalidades. Asimismo, habrá que explicitar qué variables se van a medir para conocer el grado de efectividad, y habrá que asegurarse de que éstas son relevantes para la enfermedad evaluada y que permitirán medir y cuantificar de forma fiable el grado de efectividad de los medicamentos en estudio.
2. Detallar las características de los pacientes de las cohortes, el medicamento que será evaluado en cada una de ellas y la duración del estudio. Las cohortes que se van a evaluar deberán haber sido seleccionadas una vez que el clínico haya decidido qué tratamiento va a prescribir a cada paciente (acorde con los criterios habituales de la práctica sistemática), de manera que el protocolo de estudio no modifique su hábito de prescripción. En este momento habrá que decidir qué medicamento va a ser el comparador elegido dentro de la cohorte control (y razonar la elección), así como la duración del estudio, es decir, el tiempo de seguimiento de las cohortes en evaluación. Será necesario asegurarse de que las cohortes en estudio son homogéneas en cuanto a los factores de riesgo asociados y pronósticos de la enfermedad diana. Un factor de riesgo es una variable que el investigador considera que está causalmente relacionada y es anterior al desarrollo de la enfermedad, mientras que un factor pronóstico es el que predice el curso de la enfermedad cuando ésta ya se ha iniciado. Con el fin de intentar controlarlos, es necesario introducir medidas, como el emparejamiento, la restricción y la estratificación en el momento de diseñar el estudio7. Con el fin de que las cohortes en evaluación sean lo más comparables posible, siempre habrá que intentar controlar los sesgos de selección (selection bias) y canalización (channeling bias).
El sesgo de selección se producirá cuando los pacientes de las cohortes no son comparables y su pronóstico es distinto (habitualmente presentan distintas comorbilidades asociadas y/o diferentes grados de severidad de la enfermedad evaluada), por lo que será difícil poder comparar los resultados hallados en las cohortes estudiadas. Una manera de intentar controlar este sesgo es la aplicación de técnicas de corrección del riesgo a través de modelos de ajuste de riesgo8.
El sesgo de canalización acontece cuando medicamentos con similares indicaciones terapéuticas se prescriben a grupos de pacientes con distintos factores pronósticos. Si el clínico cree que uno de los medicamentos en estudio presenta ventajas, podrá canalizar e indicar este fármaco a los pacientes con una particular comorbilidad y con mayores riesgos, y más tarde atribuir al uso de este medicamento la aparición de posteriores eventos, cuando éstos, en realidad, están en relación con las comorbilidades asociadas y el mayor riesgo de estos pacientes9.
3. Controlar adecuadamente los factores de confusión y los modificadores. Los factores de confusión y los factores modificadores son factores externos de la enfermedad que, si no se controlan de manera apropiada, pueden sesgar o distorsionar la relación entre la ingesta de un medicamento y los resultados clínicos obtenidos. Uno de los factores de confusión más frecuentes y difíciles de controlar es el que se denomina como confusión por indicación (confounding by indication), el cual aparece cuando la enfermedad que produce la indicación del medicamento actúa, a su vez, como un factor de confusión, independientemente de su gravedad10.
Para lograr un buen control será necesario recoger el máximo de datos sobre los posibles factores de confusión (toma de medicación concomitante, uso de fármacos OTC [medicamentos de dispensación sin receta), hábito tabáquico y alcohólico, hábitos de estilo de vida, etc.], aunque en muchas ocasiones será necesario recurrir a la estratificación, los análisis multivariantes y otras técnicas para poder controlar el efecto de los factores de confusión a la hora de analizar los datos de efectividad, como se verá más adelante11.
4. Cálculo del tamaño muestral necesario para que el estudio presente suficiente poder estadístico. Inicialmente será necesario fijar los errores de tipo I (error *) y de tipo II (error ß) que se van a asumir (es habitual aceptar un riesgo * ¾ 0,05 y un error ß entre 0,1 y 0,2, lo que daría al estudio un poder estadístico [1 ß] del 80-90%), y a continuación estimar la efectividad del grupo comparador, la cual habitualmente se obtiene de datos publicados en la bibliografía; en caso de que no los hubiera, habría que realizar un estudio piloto para obtenerlos.
Por último, habrá que especificar la magnitud de la diferencia del efecto que se quiere encontrar entre la efectividad lograda con los medicamentos en estudio (mínima diferencia en la efectividad que se quiere detectar), expresada en general en términos relativos, como es el riesgo relativo (RR) mínimo que se desea detectar como estadísticamente significativo. Además, siempre será necesario corregir este número en función del porcentaje de pérdidas, abandonos y retiradas de pacientes del estudio, con el fin de poder mantener su potencia estadística12.
5. Recogida de los datos de los pacientes de las cohortes, evitando el sesgo de información. Con el fin de recopilar información detallada, precisa y objetiva de los medicamentos en estudio y de los posibles factores de confusión, se recomienda diseñar cuadernos de recogida de datos (CRD) donde se recoja toda la información de interés para evaluar el grado de efectividad (medición de variables clínicas finales, medicación asociada, efectos adversos aparecidos y su tratamiento, abandonos, etc.), así como datos de posibles factores de riesgo y factores de confusión (comorbilidades asociadas, exposición a tabaco y alcohol, hábitos de vida, etc.). Todos estos datos deberían ser recogidos en todas las visitas de control que el clínico establezca de acuerdo con su práctica médica habitual, que debería ser la misma para los pacientes de ambas cohortes en estudio, con el fin de evitar cualquier sesgo de evaluación. Además, será necesario intentar medir el grado de cumplimiento terapéutico y recoger el número de abandonos en ambas cohortes, indicando las causas que lo han motivado.
6. Efectuar un seguimiento de las cohortes durante el tiempo necesario para garantizar la validez de los resultados. El tiempo de seguimiento de las cohortes dependerá de la enfermedad en estudio, los medicamentos que se quieran evaluar y el período de latencia para que se produzca el efecto terapéutico.
Habrá que estar seguro de que la duración del estudio va a ser suficiente para poder medir y valorar el efecto beneficioso de los medicamentos en estudio, y a la hora de fijarlo será deseable revisar los datos de la bibliografía y, la mayoría de las veces, consultar con clínicos expertos.
Por otra parte, será esencial que el seguimiento sea correcto e igual en todas las cohortes de pacientes en evaluación, con similares pautas de visitas, exploraciones y recogida de información.
La aparición de pérdidas de pacientes es un gran problema, ya que disminuye el poder estadístico del estudio y, además, introduce sesgos a la hora de analizar e interpretar los resultados. Por lo tanto, será importante adoptar estrategias para minimizar su aparición, tanto durante la fase de diseño y el reclutamiento de los pacientes como en la fase de seguimiento (mantener contactos periódicos con los pacientes por teléfono o correo y, en caso de que hayamos perdido la pista, llamar a familiares, amigos, médico de cabecera, etc.).
Dado que al final tendremos un porcentaje de pérdidas en cada cohorte del estudio, será obligatorio definir en el protocolo cómo se van a manejar, es decir, si se ignorarán y excluirán del análisis final, o si se emplearán técnicas de imputación con el fin de no desperdiciar la información de los pacientes perdidos13.
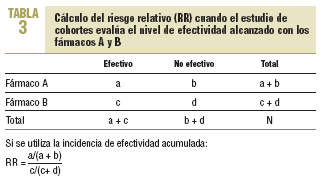
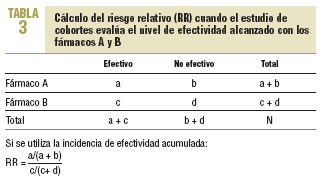
7. Análisis e interpretación de los resultados. El análisis básico en estos estudios es el cálculo de lo que se denomina como incidencia de efectividad acumulada (IEA), que es la proporción de pacientes que durante seguimiento de cada cohorte presentará el nivel de efectividad deseado tras la administración del medicamento en estudio.
Además, se obtendrá el RR (o índice de riesgo) que estimará la magnitud de la asociación entre la ingesta de un medicamento y la aparición del nivel de efectividad deseado, e indicará la probabilidad de que ésta se desarrolle en el grupo que toma un medicamento frente al otro grupo que toma el medicamento comparador (o control). Representará la razón entre la incidencia de efectividad de un grupo tratado con un medicamento y la incidencia de efectividad del grupo tratado con el medicamento comparador en el estudio.
En la tabla 3 se muestra el cálculo del riesgo relativo (RR).
A continuación es necesario calcular su intervalo de confianza (IC) al 95%, ya que el RR no es más que una estimación puntual de la verdadera magnitud de la asociación entre la exposición a los fármacos y el nivel de efectividad logrado14. Este parámetro indicará entre qué valores, con un 95% de certeza, se va a encontrar el verdadero valor de la asociación entre los medicamentos en estudio y la efectividad encontrada.
Si el RR es > 1, la probabilidad de que se produzca el nivel de efectividad deseado es mayor cuando se emplea el fármaco A frente al fármaco B, mientras que si es < 1 se produce lo contrario. Cuando el IC incluye el 1, no hay diferencias estadísticamente significativas entre el uso de un medicamento y la efectividad producida y, por tanto, no se pueden extraer conclusiones respecto de su asociación. Por otra parte, cuanto más estrecho es el IC, más precisa y fiable es la asociación encontrada15.
Aunque es posible encontrar estudios de cohortes en los que se evalúa la efectividad de 2 tratamientos mediante el empleo de la técnica de la *2 para analizar los resultados, con esta prueba estadística solamente podemos saber si hay diferencias entre las 2 cohortes, pero no la magnitud de esa diferencia. Por este motivo, es deseable y recomendable calcular el RR y su IC, ya que mostrarán claramente la cuantía de la diferencia.
Teniendo en cuenta que en un estudio de cohortes el objetivo final es conseguir una estimación no sesgada de las medidas de asociación (básicamente, el RR), el siguiente paso en su análisis es intentar controlar posibles factores de confusión o describir la modificación del efecto por terceras variables, lo que puede lograrse mediante un análisis estratificado o a través del análisis multivariante (regresión logística, regresión de Poisson y regresión de riesgos proporcionales de Cox)16.
En la actualidad se dispone, además, de otras técnicas analíticas más complejas que buscan contrarrestar los factores de confusión del estudio y que pueden emplearse si se considera que no han sido totalmente controlados con los anteriores análisis17.
Además, y con el fin de facilitar la interpretación de los resultados, es conveniente calcular y notificar el número necesario de pacientes a tratar (NNT) para producir una unidad de efectividad cuando se comparan los medicamentos en estudio, o sea, especificar el NNT del estudio. El NNT es el inverso de la reducción del riesgo relativo (1/RRA) y es un dato fácil de obtener de los resultados del estudio de cohortes; es una información que permite al agente decisor disponer de una interpretación rápida, intuitiva y relevante del resultado del estudio18.
Estudios de cohortes retrospectivas a través de bases de datos automatizadas
En estos estudios, las cohortes son elaboradas de forma retrospectiva a partir de bases de datos con las historias clínicas informatizadas. Las mayores ventajas de este tipo de diseños son la posibilidad de estudiar a un número muy elevado de pacientes a los que se ha administrado un medicamento y la obtención de resultados en cortos períodos, aunque es muy difícil manejar los sesgos y los factores de confusión, por lo que siempre será necesario ser cauto y conservador a la hora de extraer conclusiones de este tipo de diseños retrospectivos.
A la hora de su elaboración, realización y análisis, habrá que seguir los mismos principios que cuando se diseña y efectúa un estudio de cohortes prospectivo; además, hay unos puntos adicionales en los que será imprescindible incidir, como:
1. Decidir y justificar la información que se debe extraer de la base de datos. Antes de empezar el estudio, será necesario fijar detenidamente la información y los datos que será necesario obtener de la base de datos de acuerdo con el objetivo y la finalidad del estudio, como el tipo de paciente (edad, sexo, comorbilidades asociadas, etc.), el período que se desea evaluar, los medicamentos que se van a valorar, la enfermedad que se va a estudiar, etc., así como información sobre posibles variables de confusión (consumo de tabaco y alcohol, hábitos alimentarios y de estilo de vida, toma de fármacos OTC, etc.). En este punto será esencial consultar y trabajar juntos con alguien que conozca a la perfección la base de datos con la que se va a efectuar el análisis, ya que de esta manera se facilitarán mucho las cosas.
2. Identificar a los pacientes que serán incluidos en las cohortes en estudio. Habitualmente, los pacientes serán identificados porque han recibido una prescripción de los medicamentos en estudio o porque presentan la enfermedad de interés en el estudio; para ello se emplearán los códigos de la enfermedad junto con los de las pruebas complementarias más utilizadas para su diagnóstico.
A continuación, y una vez que el grupo de pacientes ha sido identificado, será necesario establecer unos criterios de selección para decidir finalmente qué pacientes serán incluidos en el estudio. Estos criterios de inclusión/exclusión deberían ser compatibles con la estructura de la base de datos, y no deberían ser más restrictivos que las contraindicaciones incluidas en la ficha técnica de los medicamentos en estudio. Además, se debería intentar que los grupos de pacientes que vayan a ser incluidos en el estudio presenten, en el momento de su inicio, unos factores pronósticos y de riesgo similares y, a poder ser, con unas comorbilidades asociadas parecidas.
Dado que el objetivo de estos estudios es conocer la efectividad de los medicamentos en condiciones de uso sistemático, sólo debería incluirse a los pacientes que han tomado los medicamentos durante el tiempo que se considere necesario para que se produzca el efecto terapéutico19.
3. Definir el episodio de tratamiento y detallar la duración del estudio. El episodio de tratamiento se refiere al período que va desde que comenzó el tratamiento con los medicamentos en evaluación hasta que finalizó, bien porque se cambió a otro medicamento o porque se dejó de tomar la medicación. Con el fin de definir episodios que sean coherentes y reflejen la práctica médica habitual, a menudo será necesario recurrir a la opinión de clínicos expertos para su definición.
La duración del estudio deberá ser lo suficientemente larga para poder medir todos los resultados clínicos relevantes y evaluar la efectividad obtenida. Se puede emplear como duración un período fijo (p. ej., 1 año de calendario o 1 año fiscal), aunque con esta metodología habrá pacientes que habrán tomado los medicamentos durante distintos períodos al haberse incorporado al estudio en diferentes etapas. Por este motivo, una segunda opción es que cada cohorte de pacientes permanezca incluida en el estudio durante el mismo período, con independencia del momento de su inclusión. El problema de esta alternativa es que hace que el estudio dure más tiempo, ya que alarga el período de inclusión de pacientes.
4. Definir cómo se van a medir y cuantificar los resultados clínicos de las cohortes en evaluación. Un reto importante cuando se emplean bases de datos para evaluar la efectividad de medicamentos es el modo de valorar la efectividad conseguida, es decir, determinar qué variables se van a medir. Lo ideal es evaluar siempre variables finales que reflejen la efectividad conseguida, que sean objetivas y que reflejen de forma fidedigna la evolución de la enfermedad estudiada.
Lo mejor es evaluar resultados que se reflejen en cambios en las analíticas o las pruebas complementarias realizadas (p. ej., cambios en el VEMS, hemogramas o en resonancias efectuadas), o bien resultados que se cuantifiquen habitualmente y que sean fáciles de medir (disminución en el número de crisis epilépticas o descensos de las cifras de presión arterial, etc.); cuando se pueda se indicarán los datos de morbimortalidad, ya que son variables finales de gran peso.
Será imprescindible intentar obtener información sobre posibles factores de confusión, especialmente el de indicación, y distintos sesgos, como el sesgo de selección (por el que el clínico asigna, de forma sistemática, un tipo de paciente a un tratamiento específico en estudio), el sesgo de la presencia de datos censurados (pacientes con distintos tiempos de seguimiento en el estudio), el sesgo del error en la medición de las variables de efectividad y el sesgo del investigador20.
5. Verificar la precisión y fiabilidad de la base de datos en la que se va a realizar el estudio. A la hora de efectuar un estudio a través de bases de datos, es esencial evaluar la fiabilidad y la validez de los datos que contiene esta fuente, ya que es muy corriente que haya imprecisiones, bien porque se han codificado mal o porque no fueron introducidos en su momento. Por tanto, será necesario revisar los siguientes puntos:
¿Hay datos perdidos y en qué porcentaje?
¿Las cohortes en estudio son seguidas de manera interrumpida durante el período de seguimiento?
¿Es posible recopilar los eventos que requirieron acudir al hospital, o bien ocurrieron cuando el paciente estuvo ingresado en el hospital?
¿Se han recogido todos los resultados de las pruebas y exámenes efectuados a los pacientes?
¿Se han auditado los datos contenidos en la base de datos a través de una revisión de las historias clínicas de los pacientes, y se ha verificado la autenticidad de la información incluida?
¿Las variables de efectividad evaluadas sucedieron durante el período de seguimiento del estudio?
Una vez revisados estos puntos, si se observa que la calidad y fiabilidad de la base de datos no son adecuadas, habría que replantearse si merece la pena continuar con el estudio, si se quiere que los resultados finales sean creíbles y válidos.
6. Discutir las limitaciones del estudio y extraer unas conclusiones lógicas y conservadoras. Cuando se realiza un estudio de cohortes retrospectivo en el seno de una base de datos, siempre hay que tener presente que en la mayoría de los casos estas bases fueron creadas con fines administrativos y de gestión, y no para efectuar estudios farmacoepidemiológicos, por lo que presentarán unas limitaciones derivadas de la falta de información y de la inclusión de posibles sesgos y factores de confusión; por ello, es necesario emplear unas técnicas analíticas especiales y ser muy precavido y conservador al extraer conclusiones de estos estudios.
Las conclusiones del estudio deberán ser coherentes y estar basadas en los resultados obtenidos. Será importante valorar si los resultados hallados son generalizables al conjunto de la población susceptible de tomar los medicamentos en estudio, y si son clínicamente relevantes, con independencia de si presentan o no significación estadística21. Además, será necesario apuntar y discutir las posibles limitaciones del estudio, de acuerdo con las deficiencias encontradas en la base de datos.
Por otra parte, es importante que se garantice a los pacientes que se va a preservar su confidencialidad y privacidad, según se indica en la legislación española y se fija en la ley de protección de datos de carácter personal.
Conclusiones
En estos momentos, cada vez es más importante disponer de datos que nos permitan conocer cómo se comportan los medicamentos en condiciones de uso habitual y qué nivel de resultados clínicos producen en la práctica médica diaria, es decir, cuál es su grado de efectividad.
Los diseños observacionales son estudios que ofrecen resultados con una validez y calidad elevadas y que son complementarios con los de los ensayos clínicos, ya que cada vez se realizan con un mejor control de los sesgos y factores de confusión y, por tanto, no se debería subestimar el potencial uso de esta clase de diseños para conocer la efectividad de los medicamentos tras su comercialización. La clave va a estar en que estos estudios intenten contestar a preguntas no conocidas y que sean de interés para la comunidad científica, y que desde luego, no induzcan a la prescripción.
Es importante que la elaboración y realización de los estudios observacionales para medir la efectividad sea de la mayor calidad posible y que éstos presenten un diseño bien elaborado; además, deben realizarse de acuerdo con la legislación vigente en la actualidad para efectuar este tipo de estudios, lo que incrementaría su uso por parte de los agentes decisores a la hora de determinar qué medicamentos deberán usarse en la práctica asistencial22-24. En este sentido, este trabajo proporciona unas recomendaciones en forma de una lista-guía que ayudará a los clínicos y otros profesionales sanitarios a la hora de diseñar, realizar y analizar correctamente estos estudios, así como a los editores y revisores de revistas biomédicas para poder valorar la validez y calidad metodológica de los estudios de efectividad enviados a las revistas para su publicación.
