





La fibrodisplasia osificante progresiva es una de las enfermedades constitucionales óseas más devastadoras, y supone un ejemplo válido para establecer el papel de la asistencia primaria en la atención a las enfermedades poco frecuentes. Aunque las enfermedades raras suelen presentar alteraciones llamativas pueden remedar síntomas y signos de trastornos comunes, con riesgo de pasar desapercibidas. Por ello, todos los profesionales sanitarios deberían proceder con un grado de sospecha razonable ante un paciente con una enfermedad aparentemente común con rasgos atípicos o evolución no convencional. En el seguimiento integral e individualizado, los cuidados dispensados por el equipo de atención primaria en coordinación con otros dispositivos asistenciales, son fundamentales. La calidad de la atención a enfermedades raras no puede ser inferior a la que se presta a los demás procesos crónicos, ya que –además de ser un imperativo de justicia y equidad– estos pacientes son, en esencia, el «paradigma de la cronicidad».
Fibrodysplasia ossificans progressiva is one of the most devastating constitutional diseases of the bone, and may be a valid example to establish the role of Primary Care in the care of rare diseases. Although rare diseases usually present with marked anomalies, they can mimic signs and symptoms of common disorders, with the risk of going unnoticed. For this reason, all health professionals should proceed with a reasonable suspicion when confronted with a patient with an apparently common disease with atypical symptoms and a non-conventional progress. The care given by the Primary Care team along with other health care services are fundamental in the integrated and individualised follow-up. The quality of care in rare diseases must not be inferior to that provided to the other chronic diseases, since, besides being a requirement of justice and fairness, these patients are, in essence, the “paradigm of chronicity”.
«This is not just a study of rare diseases or memorable cases but of exceptional patients with special needs that offered us opportunities for professional care and personal growth». «Zebras on the commons: rare conditions in family practice», W.R. Philips
Es bien conocido que las denominadas «enfermedades raras» (EERR) plantean problemas peculiares para proporcionar una atención sanitaria acorde con sus necesidades específicas1,2. A ello contribuye tanto la variedad y complejidad de estos trastornos como la dispersión de los afectados, que es inherente a los procesos de baja prevalencia. La suma de estos factores obstaculiza su investigación en cualquier faceta: básica, epidemiológica, clínica y terapéutica3,4. El retraso diagnóstico y terapéutico que conlleva2,5 está en el origen de una potencial inequidad en el acceso a los recursos y en su utilización2,6. Por ello, las EERR reciben la consideración de «desafío sanitario y social», y aparecen como una prioridad en los programas de salud de la Unión Europea1,3,7. Según su perfil clínico y evolutivo se pueden diferenciar 2 tipos: las de aparición en la edad pediátrica, casi siempre con una fuerte predisposición genética y un cuadro clínico bien definido, y aquellas de expresión clínica variable que por tener una base genética menos intensa suelen ser de aparición más tardía2.Tanto unos como otros son procesos crónicos que suelen evolucionar de forma lenta y progresiva, a menudo ocasionando limitaciones físicas importantes –incluso invalidez permanente–, por lo que pueden condicionar una notable dependencia física y psíquica, con el consiguiente deterioro de la calidad de vida2,3,7. Como consecuencia, además del sufrimiento que ocasionan a los afectados y a sus familias, las EERR consumen una parte apreciable de recursos de los sistemas de salud y repercuten por distintas vías en el conjunto de la economía2,7.
En el amplio campo de las enfermedades poco frecuentes, uno de los grupos más importantes por su repercusión en términos de frecuencia global, mortalidad y morbilidad8,9 es el de las enfermedades constitucionales del hueso (ECO)10. Este agrupa a un conjunto numeroso de entidades heterogéneas – cifrado en 456 según la última revisión del Comité internacional ad hoc—,11 con límites claros, definidos según criterios moleculares, bioquímicos o radiográficos. Además de su etiología genética (identificada con exactitud en la mayoría), el rasgo común que las caracteriza es el fracaso en alguno de los sistemas celulares del hueso, lo que causa anomalías en la transformación del tejido óseo primitivo en hueso maduro12.
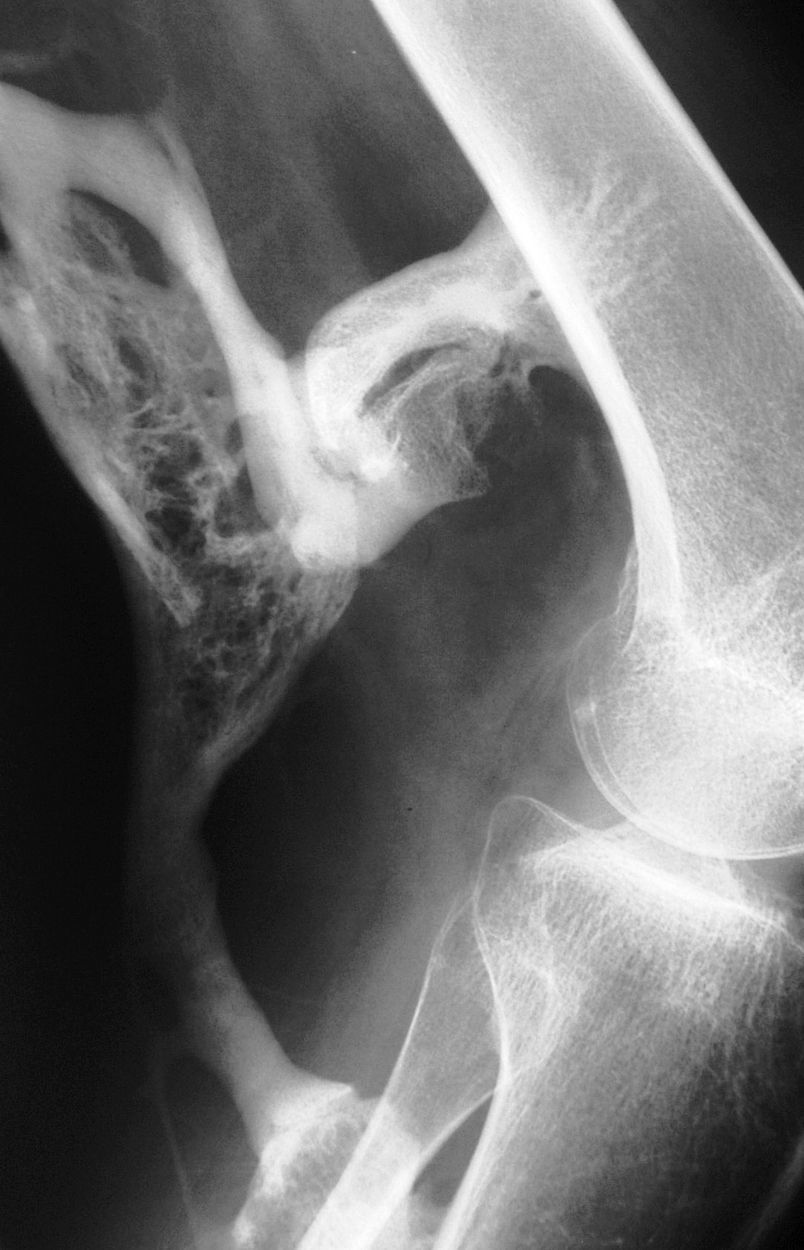
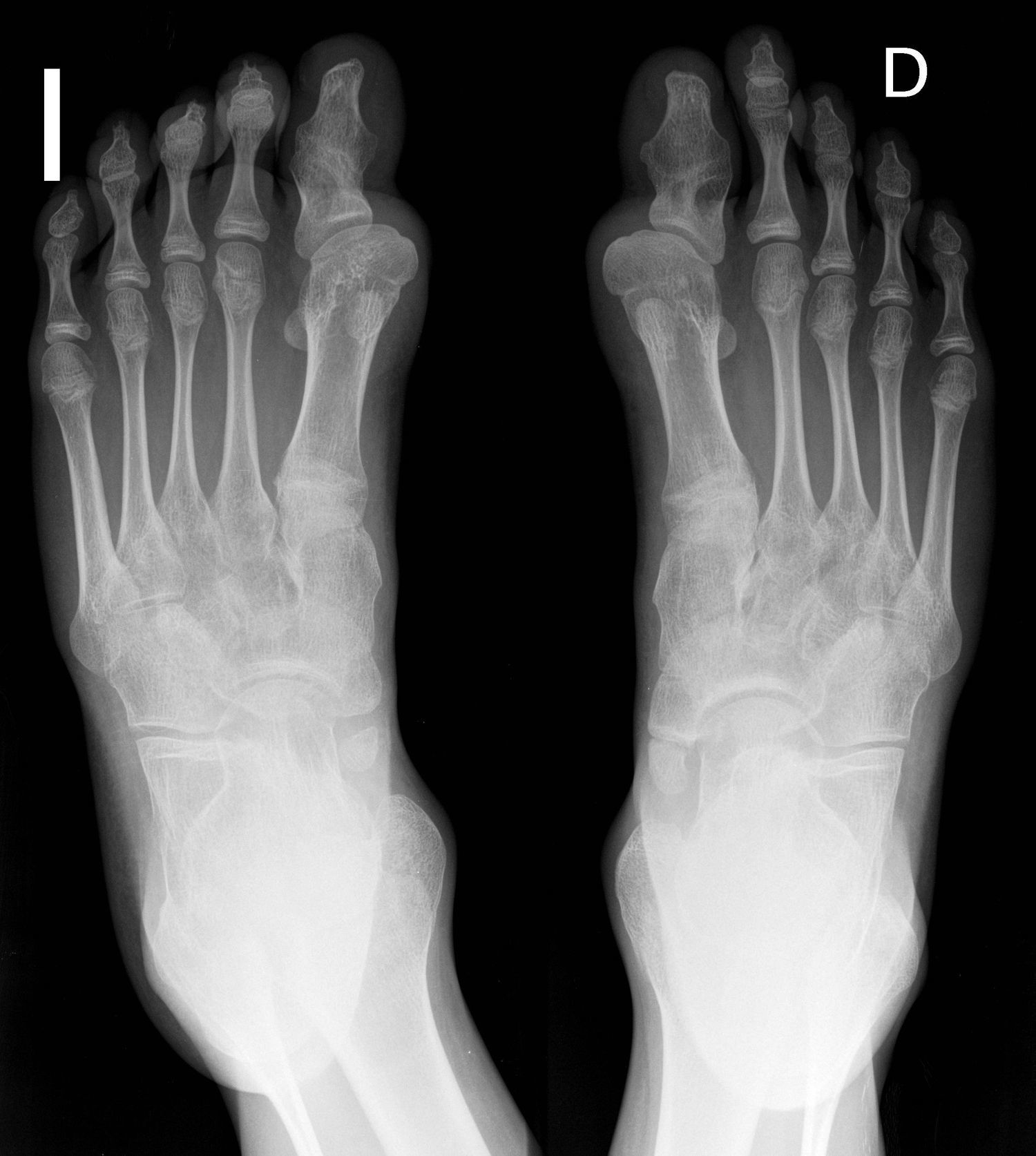
Sin duda, una de las ECO más singulares y de mayor significado es la fibrodisplasia osificante progresiva (FOP), hasta el punto de haber sido considerada «el monte Everest de los trastornos musculoesqueléticos de origen genético»13. La FOP es la causa más grave de osificación ectópica en humanos14–17. Se caracteriza por la presencia de malformaciones esqueléticas típicas (siendo el hallux valgus congénito el sello de la enfermedad) y por el desarrollo de placas de hueso maduro (según un proceso endocondral) en el seno del músculo y en otras estructuras ricas en tejido conjuntivo (osificación heterotópica)17,18. Aunque sujeta a cierta variabilidad, la formación de masas seudoinflamatorias comienza en los primeros meses o años de vida, casi siempre en relación con un traumatismo15–17. Más adelante, en la gran mayoría de los casos las placas óseas maduras continúan creciendo y ramificándose de manera incontrolable por lo que, entre la segunda y la tercera década, es habitual la pérdida de movilidad articular y una alteración funcional en todas las estructuras sobre las que asienta19. De esa forma, ciertas funciones básicas (como los movimientos respiratorios y la capacidad para ingerir alimentos) están seriamente limitadas e incluso pueden llegar a verse abolidas15–17.
En fecha reciente hemos concluido un estudio de ámbito nacional realizado en 2011 con el objetivo de evaluar la población de pacientes con FOP en España. Dada la presumible dispersión (confirmada en el curso de la investigación) y la ausencia de registros que pudieran proporcionarnos un censo fiable de casos (algo habitual en procesos de esta naturaleza20) tuvimos que recurrir a la utilización sistemática de todas las fuentes de información accesibles a fin de lograr una muestra fiable y representativa. Así, combinando la información obtenida mediante: la revisión de un archivo hospitalario de referencia en enfermedades metabólicas del hueso; los casos comunicados por la Asociación Española de Fibrodisplasia Osificante Progresiva (AEFOP), los que nos fueron notificados en respuesta a un llamamiento a las sociedades científicas, y los recuperados a través de una búsqueda bibliográfica sistemática, pudimos identificar y comprobar 24 casos con diagnóstico cierto de FOP. Dentro de esta búsqueda exhaustiva, debemos destacar la comunicación de algunos casos por parte de médicos de atención primaria en respuesta al llamamiento que realizamos a través de la Sociedad Española de Medicina de Familia y Comunitaria.
La prevalencia de FOP en todo el mundo se ha estimado en un caso por cada 2 millones de personas.1–3 Por ello nuestra cohorte representa una muestra relativamente grande ya que para una población de unos 47 millones de personas censadas en España en 2010,21 hubiera sido esperable encontrar, en el momento actual, unos 23 pacientes vivos. Las características clínicas de los 24 casos incluidos en el estudio así como sus rasgos epidemiológicos y su perfil genético han sido comunicados previamente22. Como es exigible para el diagnóstico de certeza, todos ellos habían desarrollado placas de osificación heterotópica de tipo endocondral (fig. 1) con un patrón anatómico característico y una secuencia temporal típica. Pero, para nuestro propósito, interesa destacar ahora aquellas cuestiones que ponen de manifiesto la importancia del primer nivel de la asistencia en el cuidado de estos pacientes y, por extensión, en el de otras EERR comparables. Un primer dato reseñable es que a pesar de que 21 de los 24 pacientes tenían al nacer la malformación típica del dedo gordo del pie (fig. 2) ninguno fue diagnosticado en los primeros días de vida. Además se debe mencionar el hecho de que la osificación heterotópica se manifiesta con virulencia desde los primeros meses o años, lo que condiciona que la discapacidad aparezca a una edad muy temprana. Sin embargo, la edad media en el momento del diagnóstico fue superior a los 7 años con una demora media en alcanzar el diagnóstico correcto (contando desde la aparición de la primera lesión) de casi 3 años. También es de interés práctico, por las posibilidades de prevención a partir de medidas relativamente sencillas, el que casi la mitad de los pacientes referían haber sufrido un traumatismo muscular previo al desarrollo de la lesión preósea. Pero quizá lo más llamativo es el enorme impacto que la enfermedad ocasiona en términos de limitación funcional, incapacidad y dependencia. Así, la mitad de los pacientes precisaba muletas, andador o silla de ruedas para desplazarse, y casi el 20% tenían prácticamente perdida su capacidad de deambulación, estando uno de ellos confinado en la cama desde los 25 años. Finalmente, un elevado número (cercano al 40%) presentaba una disminución importante de la audición y ninguno ha tenido descendencia.

 que caracterizan a la enfermedad, así como sinostosis de las articulaciones interfalángicas en ambos primeros dedos.")
El papel de la asistencia primaria en el cuidado a los pacientes con EERR ha recibido poca atención y la información disponible es muy escasa. La primera referencia que aborda el asunto de forma explícita es un trabajo publicado en 1993 en el Journal of Family Practice por Floyd MacIntyre, un médico de familia que ejercía en Nueva Inglaterra. En él alertaba sobre la posibilidad de ver casos «extraordinarios» en la práctica clínica, recomendando estar atentos en pacientes con una enfermedad de apariencia común pero que presenta síntomas no habituales o que sigue un curso atípico23. Años más tarde, en un trabajo de expresivo título, «Zebras on the commons: rare conditions in family practice»24, que recoge la experiencia acumulada durante 50 años en un centro de salud, se comunicó que el médico de familia es el primero en identificar una enfermedad de las denominadas «raras» en 9 de cada 10 casos, es capaz de establecer el diagnóstico definitivo en más de la mitad de ellos, y puede realizar el seguimiento completo en un número similar24. Por otra parte, en contra de lo que intuitivamente cabría pensar, la posibilidad de recibir pacientes con enfermedades raras en el centro de salud no es desdeñable. Aunque por definición se trata de trastornos que considerados uno a uno inciden en un número reducido de personas, si sumamos todos los casos presentes en los miles de procesos susceptibles de ser tipificados como «enfermedad rara», una parte importante de la población en su conjunto resulta afectada por estas enfermedades poco frecuentes («paradoja de la rareza»). Así, aunque no hay datos epidemiológicos directos, la Organización Europea para las EERR (EURORDIS) ha estimado que, en la Unión Europea, la prevalencia global de estos procesos se sitúa entre el 6 y el 8%, lo que extrapolado al conjunto de la población supone entre 24 y 36 millones de personas25, 3 millones de ellos corresponderían a España. Por otra parte, en los trastornos como la FOP que ocasionan fuertes limitaciones físicas con gran repercusión funcional y vital, la demora diagnóstica o los procedimientos inadecuados que se pudieran derivar de un diagnóstico erróneo tienen consecuencias especialmente graves2. En este sentido es muy revelador un estudio europeo sobre 8 EERR en el que el 25% de los pacientes que contestaron de forma válida referían una demora en el diagnóstico de entre 5 y 30 años5. Además, casi la mitad de los encuestados refería problemas de comunicación con el médico. Por todo ello, no resulta extraño que diversas fuentes coincidan en señalar la importancia del primer nivel de la asistencia en todas las facetas de la atención sanitaria integral y personalizada que precisan los pacientes con enfermedades poco frecuentes26–28. La cercanía al paciente y a su entorno familiar, el seguimiento (desde la niñez) continuado e inmediato, una práctica basada en la prevención y en la dispensa de cuidados, las habilidades de comunicación y el enfoque biopsicosocial tanto de la salud como de la enfermedad son circunstancias propias del primer nivel que aportan un bagaje y un abanico de posibilidades aplicables en la mejora de la atención sanitaria24,26,27. Así, la labor del médico de atención primaria podría ser esencial para detectar las alteraciones iniciales indicativas de estar en presencia de un trastorno poco común. Y, una vez orientado y tomadas las primeras medidas terapéuticas, se encuentra en una posición clave para derivar al paciente al dispositivo más adecuado para realizar el diagnóstico específico y su caracterización genética y molecular26. Pero, además, de manera coordinada con todos los demás ámbitos de la asistencia, debería seguir ejerciendo una labor central en el seguimiento de unas enfermedades que a menudo, ante la casi total ausencia de remedios eficaces, acompañan al paciente de por vida. Esto incluiría no solo atender las necesidades asistenciales en connivencia con el especialista, sino coordinar los cuidados y las acciones encaminadas a proporcionar un soporte en la esfera doméstica y social, proporcionando al paciente las ayudas que sean precisas para hacer más llevaderas las consecuencias de estos trastornos. En suma, en el centro de salud encontramos un equipo integrado por médicos, personal de enfermería, asistentes sociales y fisioterapeutas, todos ellos enfocados en torno a las necesidades del paciente y coordinados por su médico de cabecera.
Siguiendo este modelo, una iniciativa que puede ser de gran ayuda es el denominado protocolo DICE de atención primaria en enfermedades raras (DICE-APER). Se trata de una herramienta de trabajo online elaborada conjuntamente por el grupo de trabajo de enfermedades raras de la Sociedad Española de Medicina Familiar y Comunitaria (SEMFyC) y el Instituto de Investigación en Enfermedades Raras (IIER), en colaboración con la Federación Española de Enfermedades Raras (FEDER) y el Centro Estatal de Enfermedades Raras (CREER). Recoge en sus siglas las iniciales de los 4 elementos básicos que debe incluir la atención sanitaria de las EERR: «D»(diagnóstico), «I» (información sobre la enfermedad y los recursos disponibles), «C» (coordinación de la asistencia y canales de comunicación con el especialista, y «E» (epidemiología: información al sistema sanitario). Su sencilla aplicación y la posibilidad de hacer un enfoque global aportan elementos de gran interés cuya utilidad está siendo evaluada.
Sin embargo, la realidad de la atención primaria en España, con una fuerte carga asistencial24 y unos profesionales cuya formación específica en este terreno es generalmente escasa26, dificulta proveer la atención sanitaria de calidad que demandan estos pacientes. A ello contribuye también el hecho de que, por su gran diversidad y complejidad, la atención sanitaria de los procesos de baja frecuencia requiere un enfoque multidisciplinario y unos conocimientos y habilidades específicas por parte de los profesionales2. Sin embargo, en la actualidad la oferta de formación en EERR es insuficiente (aunque se detectan grandes diferencias territoriales) y poco coordinada28. Profundizando en esta carencia, los datos del «Estudio cualitativo AP40» indican que el déficit de conocimiento de las EERR en el ámbito de la asistencia primaria es común a los distintos profesionales (médicos, enfermeras y trabajadores sociales) e independiente de si desarrollaran su labor en el medio rural o urbano29. Si a todo ello añadimos la dificultad de acceso a ciertos procedimientos de diagnóstico y los problemas de coordinación entre la atención primaria y la hospitalaria24,26,27 es comprensible que los pacientes con EERR y sus familias deban hacer frente a problemas como «falta de acceso a un diagnóstico correcto, déficit de conocimiento, consecuencias sociales indeseables, y falta de equidad en la asistencia sanitaria»1. Resulta esperanzador que fuentes de distinta procedencia señalen que la situación puede ser mejorada mediante un abordaje sistemático que tenga en cuenta la perspectiva de la atención primaria y su coordinación con otros ámbitos asistenciales y sociales1,26,27.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Nuestro mayor reconocimiento y aprecio a los miembros de la Asociación Española de Fibrodisplasia Osificante Progresiva, en particular a Patricia Martín Murciano, por su generosa y entusiasta colaboración. Agradecemos a María Teresa Mombiela Muruzabal, especialista en Nefrología y Medicina de Familia y Comunitaria (Centro de Salud de Monte Rozas) su experta valoración del manuscrito y sus comentarios.
