







Introducción
Desde mediados de la década de los treinta se sabe que la enfermedad celíaca (EC) es más frecuente entre los familiares de pacientes diagnosticados1,2. Actualmente se estima que la prevalencia de la EC en la población general europea se sitúa en torno al 1%3, mientras que en familiares de primer grado los porcentajes oscilan entre el 2,6 y el 16%4,5. Recientemente, en Estados Unidos, Fassano et al6 han encontrado una prevalencia del 4,5%, intermedia a las registradas en nuestro país del 2,2-10%7,8. Sin embargo, difícilmente estos familiares son diagnosticados por la clínica, ya que el 50% son asintomáticos o presentan formas atípicas que dificultan su identificación9.
Con el diagnóstico precoz de la EC y el inicio de la dieta sin gluten mejora la calidad de vida del paciente, se evita el desarrollo futuro de complicaciones10 y enfermedades asociadas de naturaleza autoinmune, como el linfoma intestinal11 y la diabetes mellitus tipo 112, además de reducirse la mortalidad13 y disminuir los costes económicos originados en consultas y pruebas complementarias, realizadas durante años, hasta que se obtiene el diagnóstico de EC14.
El objetivo del estudio ha sido investigar la presencia de EC entre los familiares de primer grado de pacientes celíacos, así como las posibles diferencias clínicas entre familiares diagnosticados y sus respectivos casos índices.
Material y métodos
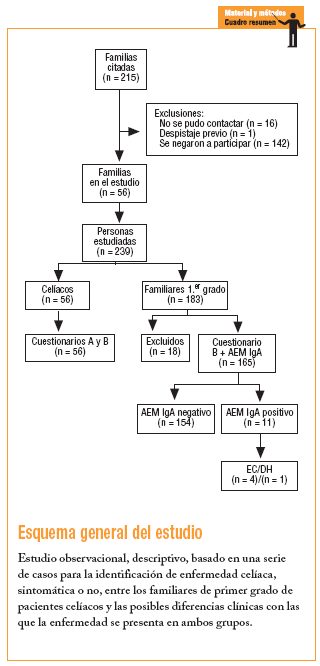
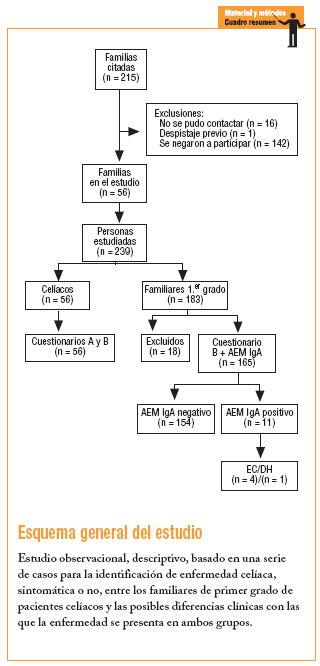
Durante los años 2001 y 2002, a las 215 familias de Sevilla con un paciente celíaco inscrito a finales del 2000 en la Asociación de Celíacos de Andalucía, se les remitió una carta informativa; después, en una entrevista telefónica, se ofreció la posibilidad de participar en el estudio a todos los familiares de primer grado a los que no se les hubiese realizado el cribado de la EC.
Fue imposible contactar con 16 familias. De las 199 restantes, sólo en uno se había realizado previamente el cribado a todos los miembros (padre, madre y 2 hermanos); 142 se negaron a participar a pesar de contar con 355 familiares de primer grado, de los que sólo habían sido estudiados 21 hermanos (5,9%) de 9 pacientes, y 56 aceptaron participar. Dichas familias fueron citadas a la consulta, donde se comprobó la existencia de un miembro celíaco en cada una de ellas y de 183 familiares de primer grado, con un número total de 239 participantes que firmaron el correspondiente consentimiento informado.
Los 56 pacientes o casos índice cumplimentaron los cuestionarios A, donde se registraron datos sobre su EC, y B, en el que se recogió información sobre la filiación, las enfermedades actuales, y los antecedentes personales y familiares.
En cuanto a los familiares, a 18 no se les pudo realizar la determinación del anticuerpo antiendomisio IgA (AEM IgA), por lo que quedaron excluidos del estudio. Los 165 restantes cumplimentaron el cuestionario B y se les solicitó el marcador serológico referido. Los casos con resultados positivos fueron derivados a sus médicos de cabecera con un informe sobre el análisis realizado y la necesidad de proseguir el estudio mediante la repetición de la analítica y/o la derivación al gastroenterólogo.
Los AEM IgA de la primera determinación se realizaron mediante inmunofluorescencia indirecta, usando como sustrato la porción distal de esófago de mono verde africano, de acuerdo con el método descrito por Chorzelsky et al15. Los resultados fueron considerados positivos cuando el patrón reticular inmunofluorescente fue observado en la muscularis mucosae a una dilución de suero >= 1:5. La validez diagnóstica de los AEM IgA en uno de los estudios con mayor número de familiares investigados para la detección de casos, realizado en España, tuvo una sensibilidad (S) del 100%, una especificidad (E) del 99,6%, un valor predictivo positivo (VPP) del 89,4% y un valor predictivo negativo (VPN) del 100%16. Recientemente, en Italia se han obtenido valores de S del 100%, E del 100%, VPP del 100% y VPN del 100% en pacientes sintomáticos17. La toma de la biopsia, salvo en 1 caso en el que se obtuvo con cápsula de Watson, se realizó mediante fibroduodenoscopia. Para establecer el diagnóstico se emplearon los criterios definidos por la Sociedad Europea de Gastroenterología y Nutrición Pediátrica en 1969, revisados posteriormente en 198918,19. El estudio anatomopatológico se realizó según la clasificación de Marsh20.
Desde el punto de vista clínico, los pacientes fueron clasificados como «asintomáticos» cuando no presentaron síntomas, «sintomáticos atípicos» cuando tenían manifestaciones gastrointestinales no hipoabsortivas o bien éstas eran extraintestinales, y «sintomáticos típicos» cuando la manifestación inicial fue el síndrome clásico de hipoabsorción. La «dermatitis herpetiforme» (DH), considerada una manifestación de la intolerancia al gluten, constituye, junto con la EC, la enteropatía sensible al gluten. Para su diagnóstico se realizó el estudio anatomopatológico cutáneo perilesional, mediante inmunofluorescencia directa, en el que se puso de manifiesto la presencia de los depósitos granulares de IgA específicos en la unión dermoepidérmica.
Las variables cualitativas se expresaron en forma de proporciones, las variables cuantitativas como media ± desviación estándar (DE), las comparaciones de medias entre grupos se realizaron mediante el test de la t de Student-Fisher, una vez comprobada la normalidad de la variable mediante el test de Kolmogorov-Smirnov.
Resultados
De los 165 familiares estudiados, 11 presentaron AEM IgA positivos (6,6%) y todos fueron derivados al gastroenterólogo por sus médicos de cabecera. Sólamente 5 accedieron al estudio endoscópico para la obtención de la biopsia intestinal, confirmándose el diagnóstico de EC en 4 y DH en uno (3%); 4 (80%) eran hermanos y uno (20%) hijo de sus respectivos casos índices. No se detectó ningún caso entre los padres (tabla 1). Los 6 familiares restantes (54,5%) no quisieron realizarse la endoscopia argumentando que no tenían síntomas de EC (tabla 2). No obstante, en el caso 5, al persistir la anemia, los dolores abdominales y la serología positiva, con posterioridad al estudio se confirmó el diagnóstico de EC. En el caso 11, que presentaba anticuerpos positivos, se mantuvieron los dolores abdominales y las cefaleas, a pesar de lo cual el paciente no accedió al estudio endoscópico. En los otros 4 no se produjo ningún cambio en su situación clínica.

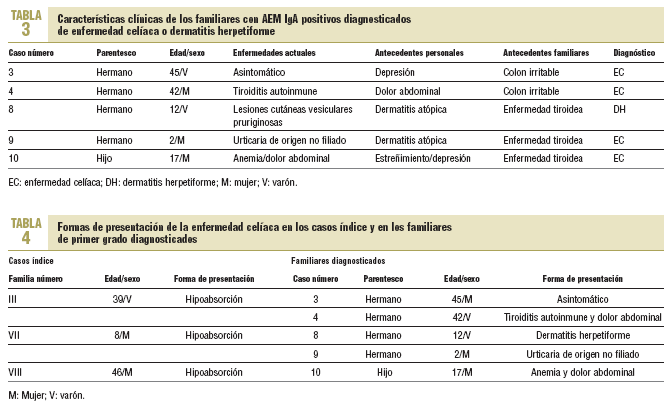
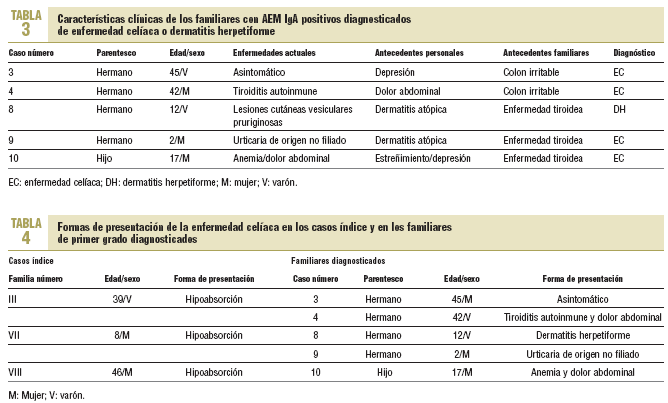
La edad media de los familiares en el momento del diagnóstico fue de 23,6 años (rango, 2-45). Solamente uno, con antecedente de depresión mayor, estaba asintomático (caso 3). Los otros habían sido diagnosticados de tiroiditis autoinmune y dolor abdominal no filiado (caso 4), dermatitis atópica con la demostración posterior de que se trataba de una DH (caso 8), urticaria no filiada junto a dermatitis atópica (caso 9) y anemia ferropénica resistente a tratamiento con dolores abdominales inespecíficos (caso 10) (tabla 3). No hubo diferencias entre los que accedieron o no a la biopsia para variables como la edad media, los antecedentes personales y familiares o las enfermedades actuales.
En los casos índice (un varón y 2 mujeres pertenecientes a las familias III, VII, VIII), la edad media en el momento del diagnóstico fue de 31 años (rango, 8-46 años) y la forma de manifestación, el síndrome de hipoabsorción. Se comprobó la presencia de agregación en las familias III y VII (tabla 4).
Discusión
En este estudio, las tasas de familiares con AEM IgA positivo (6,6%) se sitúan en cifras intermedias entre las registradas en el País Vasco por Vitoria et al16 (2,6%) y las halladas en Finlandia por Mäki et al5 (12%). En cuanto al número de casos diagnosticados (3%), su cifra está por encima de las halladas por López Hoyos et al7 (2,2%) y Vitoria et al16 (2,9%) en España, y por debajo de las registradas por Mustalahti et al4 (6,2%) en Finlandia y Korponay-Szabó et al21 (8,5%)en Hungría.
Al igual que en otras investigaciones7, el escaso número de biopsias efectuadas (45,4%) origina un distanciamiento de las cifras reales de EC en este colectivo. No obstante, al comprobarse que todos los familiares con AEM IgA positivos presentaron síntomas y/o antecedentes personales o familiares compatibles con la EC (datos no mostrados), así como una elevada concordancia diagnóstica entre los resultados serológicos y anatomopatológicos (tabla 3), se sospechó que el número de casos de EC hubiese sido más elevado con un mayor número de familiares biopsiados, como pudo comprobarse posteriormente en el familiar 5 y quizás en el 11. A ello habría que añadir la ausencia de falsos positivos entre los familiares de primer grado de casos diagnosticados, según sugieren Vázquez et al22. La poca información sobre la prevalencia de la EC y sus formas de manifestarse entre los propios familiares23, además de la ausencia de sintomatología clásica24 y, a veces, la falta de insistencia de los médicos en la necesidad de excluir el diagnóstico en dicho grupo de riesgo, pudieron justificar el rechazo al estudio endoscópico.
No se efectuó la determinación de la inmunoglobulina A (IgA), lo que pudo interferir en los resultados del marcador serológico, dado que al ser los déficit de IgA 10 veces más frecuentes en los pacientes celíacos, su falta de diagnóstico pudo originar falsos negativos25.
En el presente trabajo también se constata, por una parte, que la presentación clínica de los 5 casos diagnosticados dista de ser la típica, con mayor frecuencia de formas asintomáticas (un caso)26,27, atípicas (3 casos)28,29 ydermatitis herpetiforme (un caso)30, y por otra, el retraso diagnóstico, ya que la edad media fue de 23,6 años.
Cuando la EC se manifiesta, como ocurre en los casos índice, con el síndrome de hipoabsorción, el diagnóstico suele establecerse con facilidad. Sin embargo, la edad media en el momento del diagnóstico fue superior en 7 años a la presentada por los familiares diagnosticados. Este retraso resulta llamativo si tenemos en cuenta la clínica y los antecedentes registrados, puesto que en ellos se aprecia la presencia de síntomas relacionados con la EC antes de establecerse el diagnóstico definitivo. Así, el caso índice de la familia III presentaba dolores abdominales diagnosticados de meteorismo inespecífico desde los 13 años; y el de la familia VIII se trataba de una paciente de talla baja, con anemia ferropénica resistente a tratamiento de origen no filiado, diagnosticada de colon irritable desde los 33 años. Circunstancias parecidas se ponen de manifiesto en los antecedentes personales de los familiares diagnosticados en el estudio (tabla 3).
Es evidente que el retraso diagnóstico de la EC está relacionado con la mayor o menor similitud entre sus síntomas de presentación y el síndrome hipoabsortivo. Actualmente, está demostrado que la sintomatología gastrointestinal típica depende de la superficie intestinal lesionada31. Cuando el intestino indemne suple el déficit absortivo de la zona afectada, la sintomatología gastrointestinal característica no se hace patente. A medida que la enfermedad va incluyendo zonas cada vez más amplias de duodeno y yeyuno, los síntomas se van asemejando progresivamente al cuadro característico y nos permiten establecer el, hasta entonces, complicado diagnóstico.

No obstante, son necesarias más investigaciones sobre las formas clínicas de presentación de la EC, sus características serológicas e histológicas, así como la realización de estudios de coste-efectividad en los que apoyar la recomendación del cribado en familiares.
En conclusión, la mayor frecuencia de EC encontrada entre los familiares de primer grado, junto con su presentación clínica asintomática o atípica, justifica la búsqueda activa de casos. Este colectivo, en el momento del diagnóstico, debería ser mejor informado sobre la necesidad de realizar un cribado de la EC como estrategia para aumentar el porcentaje de aceptación de éste.
