





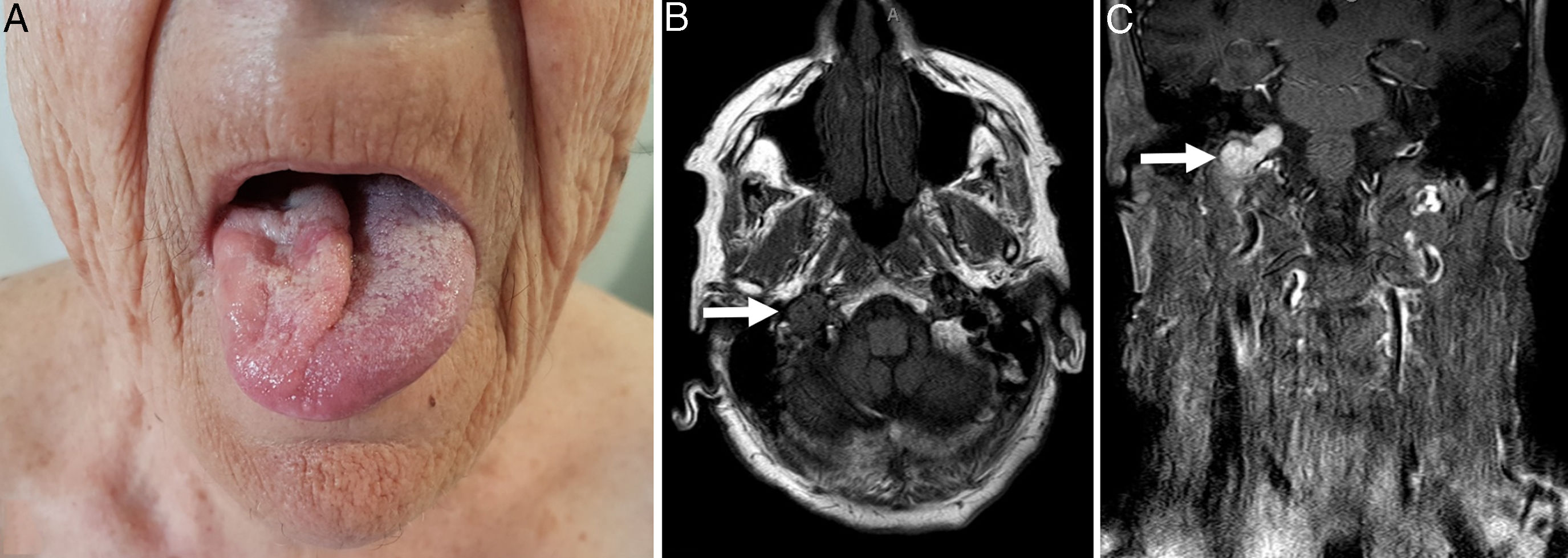
An 80-year-old woman with chronic ischemic heart disease presented for evaluation of a 6-months history of dysphonia. The patient did not have previous surgery or radiation, and did not relate headache, loss of audition or loss of weight. Physical examination revealed hemiatrophy of the tongue, on the right side, and the inability to completely deviate the tongue toward the left side of the mouth on protrusion (Fig. 1A) and the remainder was unremarkable. Laryngoscopic examination showed paralysis of the right vocal cord. Laboratory tests including biochemical, immunological, and microbiological profiles were normal. T1-weighted magnetic resonance imaging (MRI) demonstrated a 35×20×20mm hypointense tumor occupying the right jugular foramen and extending into the hypoglossal canal (Fig. 1B). The tumor enhanced intensely after gadolinium injection and showed a characteristic “salt-and-pepper appearance” (Fig. 1C). Right vagal and hypoglossal nerve damage caused denervation atrophy of the tongue and paralysis of the right vocal cord. The right jugular vein and carotid artery appeared intact. The most probable diagnosis was jugular paraganglioma (JP). Catecholamines in 24-h urine were not increased and a computer tomography (CT) of the thorax, abdomen and pelvis ruled out other tumor foci. Due to the high surgical risk, conservative treatment with periodic follow-up visits every 3 months was selected. Currently, 6 months after diagnosis, the clinical status remains stable and no other manifestations have appeared.

A: Hemiatrophy of the tongue, on the right side, and the inability to completely deviate the tongue toward the left side of the mouth on protrusion. B: T1-weighted MRI demonstrated a 35×20×20mm hypointense tumor occupying the right jugular foramen and extending into the hypoglossal canal. C: The tumor enhanced intensely after gadolinium injection and showed a characteristic salt-and-pepper appearance.
Paragangliomas are rare, highly vascular neoplasms that arise from specialized neural crest chromaffin cells. 30% of them appear as part of hereditary syndromes. Paragangliomas can derive from either parasympathetic or sympathetic ganglia. As in our patient, the former are located almost exclusively in the head and neck.1 The most common paraganglioma locations of the head and neck in descending order are the carotid body, jugular, tympanic, and vagal paragangliomas.2 Less than 4% of paragangliomas secrete catecholamines.1 The estimated incidence of paragangliomas of the head and neck is 1:30,000. Though histologically benign, they can be locally aggressive by invading adjacent structures. The clinical presentation of a paraganglioma varies based on the tumor location. JP often present with pulsatile tinnitus and hearing loss and frequently cause dysfunction of the cranial nerves VII–XII with large tumors. Along with medical history and targeted physical examination, imaging is essential for its diagnosis and management. The tumor appears hypointense on T1-weighted images, isointense/hyperintense on T2-weighted images, and enhances intensely after gadolinium injection on MRI. Signal voids related to the high flow in the tumor are common with a “salt-and-pepper appearance”. The main imaging differential diagnosis for a JP includes metastases, meningioma, hemangiomas, Paget disease, melanoma, middle ear adenoma, acoustic neuroma, and an endolymphatic sac tumor.2 Treatment should be individualized based on the symptomatology, size, and location of the tumor. The different treatment options include embolization, surgical excision, conventional radiotherapy, and stereotactic radiosurgery.2,3 In selected cases, a watchful approach with repeat imaging may be warranted.2
