


Los aerosoles orgánicos secundarios (AOS) constituyen una fracción de las partículas atmosféricas. Este tipo de partículas se forman como consecuencia de la reacción de oxidación de ciertos gases orgánicos, lo que conduce a la formación de compuestos de baja volatilidad. Del mismo modo que para otros contaminantes atmosféricos, los modelos de calidad del aire permiten la simulación de partículas, una herramienta muy útil en tareas de gestión de la calidad del aire. Sin embargo, el uso adecuado de estos modelos debe basarse en la validación de su capacidad para reproducir las concentraciones observadas. Las estaciones de monitoreo de la calidad del aire registran información sobre una amplia variedad de contaminantes atmosféricos. Desafortunadamente, no se dispone habitualmente de medidas de AOS, ya que la instrumentación que se tiene en dichas redes de monitoreo no permite la diferenciación de las fuentes primarias y secundarias de los aerosoles orgánicos. Este documento presenta una serie de experimentos de fotooxidación realizados en las cámaras de simulación del Fotorreactor Europeo (CEAM, España) en diferentes condiciones experimentales con objeto de obtener datos sobre la formación de AOS. El uso de este tipo de cámaras permite aislar los procesos químicos y de formación de aerosoles, por lo que los datos presentados en este estudio tienen un considerable valor para propósitos de evaluación de modelos de formación de AOS, al igual que para el estudio del comportamiento de este tipo de partículas.
Secondary organic aerosols (SOA) constitute a significant fraction of the atmospheric particulate matter. Theses particles are formed as a consequence of the oxidation reaction of certain organic gases that leads to the formation of low-volatility compounds. As for other pollutants, air quality models allow the simulation of particle levels and thus models constitute a powerful tool in air quality management. Nevertheless, the accepted use of models must be based on the validation of its capacity to reproduce observed concentrations. Air monitoring sites provide measured information of a large variety of ambient pollutants. Unfortunately, measurements on SOA are not normally available, as current monitoring networks do not include instrumentation to distinguish primary from secondary sources of organic carbonaceous aerosol. This paper presents a set of photooxidation experiments performed in the European Photorreactor (EUPHORE) smog chamber (CEAM, Spain) under different experimental conditions to investigate SOA formation. The use of chambers allows the isolation of atmospheric chemistry and aerosol formation processes. Thus, although these measurements were obtained at initial precursor concentrations higher than those in atmospheric conditions, they constitute a valuable set of information for SOA model evaluation purposes.
Atmospheric particles can have a significant impact on human health. High particle concentration levels, especially of the fine fraction, can negatively affect human health and ecosystems. Because of this, clean air legislation all around the world aims at avoiding high atmospheric particle levels by establishing air quality thresholds that should not be exceeded (EU, 1999; EU, 2008).
A wide range of inorganic and organic compounds can be present in particulate matter. Organic compounds make up a substantial fraction of atmospheric fine particulate matter, accounting for 20-90% of aerosol mass in the lower troposphere (Kanakidou et al., 2005). Both inorganic and organic atmospheric particles can be directly emitted by anthropogenic (traffic and power plants emissions) or natural sources (such as volcanoes or dust storms). They can also be formed in the atmosphere as a consequence of chemical and physical processes (secondary particles). Organic particles formed in the atmosphere are commonly referred to as secondary organic aerosols (SOA).
Although there are very few studies focused on the effects of SOA on health, the fact that summer high particle levels are commonly associated with the production of SOA could indicate a contribution of SOA to the association between daily mortality and fine particles observed in summer periods, as suggested in Alfaro-Moreno et al. (2007).
To simulate particle mass concentrations all the processes affecting atmospheric particle levels must be considered, and thus SOA formation must be included in model codes. Knowledge of the processes that involve SOA formation has significantly increased over the last few years (Hallquist et al., 2009). The most studied and probably the most important mechanism of SOA formation is the oxidation of volatile organic compounds (VOCs), frequently produced through the reaction with OH radicals, forming products of lower volatility that subsequently partition into the condensed phase (Kroll and Seinfeld, 2008). Additional pathways consisting of the reaction of less-volatile organic gases may also lead to the production of SOA (Robinson et al., 2007).
Both biogenic and anthropogenic sources of VOCs contribute to SOA formation (Kanakidou et al., 2005). Until recently, the major biogenic SOA precursors were believed to be the terpenes, being a-pinene and limonene some of the most important ones (Griffin et al., 1999; Kanakidou et al., 2005). However, the importance of isoprene as a biogenic SOA precursor has been confirmed in a number of recent field and laboratory experiments (Claeys et al., 2004; Kleindienst et al., 2006). Whilst biogenic VOCs are the dominant contributors to global SOA formation, anthropogenic species, such as aromatic compounds (benzene, toluene, xylenes and trimethylbenzenes), can contribute significantly to the production of SOA in urban areas (Kanakidou et al., 2005).
The oxidation processes of different VOCs have been studied in depth in chambers. These experiments offer a framework for the study of chemical reactions under controlled conditions, and thus constitute a very useful tool to investigate how specific processes involved in SOA formation take place. In most of the previous experiments, the formation of SOA was investigated by introducing one single VOC or binary mixtures. Lim and Ziemann (2005) analyzed the alkane oxidation with the OH radical in the presence of NOx. For some aromatic compounds, such as 1,3,5-trimethylbenzene (1,3,5-TMB), toluene, and xylenes, the SOA formation potential has also been investigated (Kalberer et al., 2004; Song et al., 2005; Weitkamp et al., 2007). Regarding biogenically-emitted VOCs, experiments based on a-pinene and limonene have also been presented in a large number of publications (Kamens and Jaoui, 2001; Leungsakul et al., 2005; Yu et al., 2008). Some research has also been focused on the role of isoprene on SOA formation (Claeys et al., 2004; Kroll et al., 2006). Experiments performed by introducing diesel and gasoline in chambers have also been presented (Odum et al., 1997; Weitkamp et al., 2007). Nevertheless, investigating the role of the individual VOCs on SOA formation is difficult when so many gases are present.
A recent publication on the current and emerging issues related to SOA suggests that more complex mixtures than just one or two VOCs should be investigated in laboratories studies, in order to describe their effect on SOA formation (Hallquist et al., 2009). In Vivanco et al. (2011) we presented the results of ten photooxidation experiments performed in the European Photorreactor (EUPHORE) outdoor chamber (CEAM, Valencia, Spain) in order to study SOA formation under different VOCs initial conditions. Those experiments were performed by introducing a mixture of toluene (TOL), 1,3,5 trimethylbenzene (TMB), o-xylene (OXYL), octane (OCT) and mixtures of isoprene (ISO), limonene (LIM) and a-pinene (APIN) into the EUPHORE photoreactor. In this paper the results of 23 new experiments performed in 2009, 2010 and 2011 again in the EUPHORE photoreactor are presented. Further relative humidity, oxidant and acidic conditions were explored. Also two experiments with an even more complex mixture were performed, by introducing all the mentioned anthropogenic and biogenic VOCs and benzene. With these new experiments the aim has been to facilitate the evaluation of air quality models, since normally no SOA information is available due to the difficulty of distinguishing between primary and secondary components. Also the isolation of the chemistry and aerosol formation in chambers facilitates the evaluation of these specific processes, which is not possible when comparing to real atmosphere observations, as other atmospheric processes are involved.
2Experimental section2.1Experimental conditionsThe EUPHORE photoreactor is a half-spherical fluorine-ethene-propene (FEP) foil chamber mounted on an aluminum floor covered with FEP foil. Teflon is transparent to solar radiation and more than 80% light transmission is reached in the range of 280-640nm (range at which photochemical atmospheric processes take place). The chamber volume is approximately 200 m3. Two mixing fans are used to ensure homogeneity of the reaction mixtures. Analysis equipment is located below the aluminum floor. The EUPHORE facility has been described in detail elsewhere (Becker, 1996; Volkamer et al., 2001)
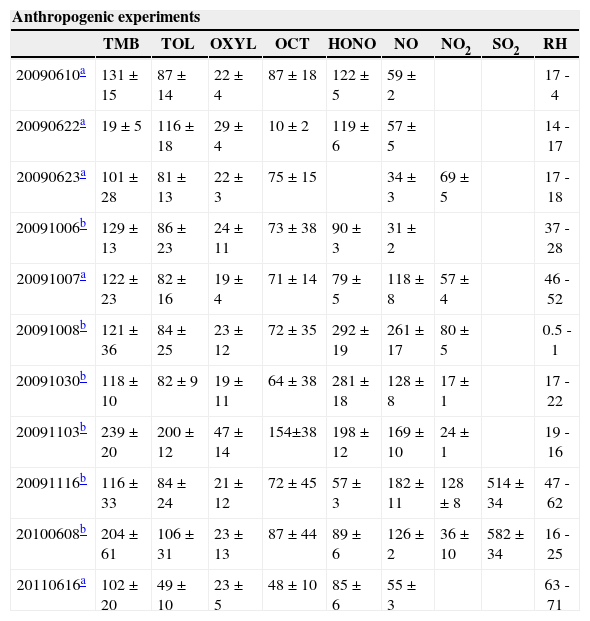
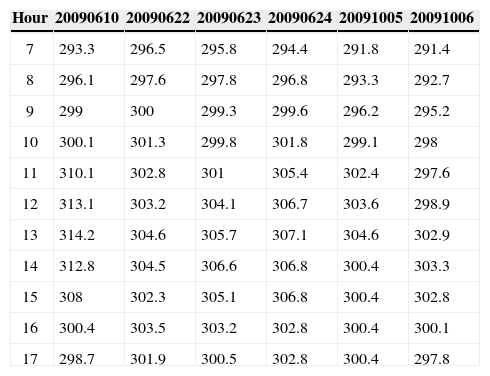
A total of 23 experiments were performed in the EUPHORE smog chamber. They were carried out in June, October and November 2009, June 2010 and June 2011. Eleven experiments were carried out using mixtures of anthropogenic VOCs (1,3,5 trimethylbenzene [TMB], o-xylene [OXYL], octane [OCT], and toluene [TOL]) and ten with mixtures containing biogenic VOCs (a-pinene [APIN], limonene [LIM], and isoprene [ISO]). Two experiments with a more complex mixture of some anthropogenic and biogenic parent VOCs were also performed. Initial concentrations and their uncertainty range, as well as other experimental conditions are presented in Table I. Temperature registered during the experiments is presented in Table II.
Initial conditions for the new set of experiments. Concentrations are given in ppb and relative humidity (RH) ranges in %. The ranges for RH indicate values at the starting and ending time.
| Anthropogenic experiments | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| TMB | TOL | OXYL | OCT | HONO | NO | NO2 | SO2 | RH | |
| 20090610a | 131 ± 15 | 87 ± 14 | 22 ± 4 | 87 ± 18 | 122 ± 5 | 59 ± 2 | 17 - 4 | ||
| 20090622a | 19 ± 5 | 116 ± 18 | 29 ± 4 | 10 ± 2 | 119 ± 6 | 57 ± 5 | 14 - 17 | ||
| 20090623a | 101 ± 28 | 81 ± 13 | 22 ± 3 | 75 ± 15 | 34 ± 3 | 69 ± 5 | 17 - 18 | ||
| 20091006b | 129 ± 13 | 86 ± 23 | 24 ± 11 | 73 ± 38 | 90 ± 3 | 31 ± 2 | 37 - 28 | ||
| 20091007a | 122 ± 23 | 82 ± 16 | 19 ± 4 | 71 ± 14 | 79 ± 5 | 118 ± 8 | 57 ± 4 | 46 - 52 | |
| 20091008b | 121 ± 36 | 84 ± 25 | 23 ± 12 | 72 ± 35 | 292 ± 19 | 261 ± 17 | 80 ± 5 | 0.5 - 1 | |
| 20091030b | 118 ± 10 | 82 ± 9 | 19 ± 11 | 64 ± 38 | 281 ± 18 | 128 ± 8 | 17 ± 1 | 17 - 22 | |
| 20091103b | 239 ± 20 | 200 ± 12 | 47 ± 14 | 154±38 | 198 ± 12 | 169 ± 10 | 24 ± 1 | 19 - 16 | |
| 20091116b | 116 ± 33 | 84 ± 24 | 21 ± 12 | 72 ± 45 | 57 ± 3 | 182 ± 11 | 128 ± 8 | 514 ± 34 | 47 - 62 |
| 20100608b | 204 ± 61 | 106 ± 31 | 23 ± 13 | 87 ± 44 | 89 ± 6 | 126 ± 2 | 36 ± 10 | 582 ± 34 | 16 - 25 |
| 20110616a | 102 ± 20 | 49 ± 10 | 23 ± 5 | 48 ± 10 | 85 ± 6 | 55 ± 3 | 63 - 71 | ||
| Biogenic experiments | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| ISO | APIN | LIMO | HONO | NO | NO2 | SO2 | RH | ||
| 20090624c | 107 ± 3 | 66 ± 5 | 58 ± 4 | 99 ± 6 | 34 ± 2 | 128 ± 1 | 0.5 - 3 | ||
| 20091005a | 121 ± 34 | 64 ± 18 | 56 ± 16 | 43 ± 3 | 26 ± 2 | 17 - 11 | |||
| 20091014c | 92 ± 3 | 50 ± 5 | 50 ± 4 | 87 ± 5 | 48 3 | 30 - 26 | |||
| 20091026b | 122 ± 33 | 71 ± 18 | 40 ± 10 | 53 ± 3 | 41 ± 2 | 19 - 22 | |||
| 20091027a | 63 ± 18 | 65 ± 18 | 101 ± 7 | 32 ± 2 | 8 - 13 | ||||
| 20091029b | 99 ± 28 | 59 ± 12 | 53 ± 13 | 307 ± 20 | 150 ± 10 | 8 - 11 | |||
| 20091111c | 87 ± 3 | 50 ± 5 | 51 ± 4 | 40 ± 3 | 244 ± 15 | 89 ± 5 | 513 ± 34 | 19 - 19 | |
| 20100607c | 88 ± 3 | 79 ± 5 | 76 ± 4 | 165 ± 10 | 198 ± 1 | 461 ± 34 | 20 - 21 | ||
| 20110617a | 73 ± 15 | 40 ± 8 | 76 ± 15 | 78 ± 8 | 47 ± 3 | 45 - 70 | |||
| 20110621a | 67 ± 13 | 26 ± 5 | 68 ± 14 | 91 ± 6 | 59 ± 4 | 13 ± 1 | 48 - 63 | ||
| Mixed experiments | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| TMB | TOL | OXYL | OCT | HONO | NO | NO2 | SO2 | RH | |
| 20091106b, c | 30 ± 7 | 167 ± 20 | 39 ± 11 | 25 ± 7 | 105 ± 7 | 141 ± 9 | 59 ± 4 | 0.4 - 3 | |
| BENZ | ISO | APIN | LIMO | ||||||
| 39 ± 11 | 8 ± 3 | 17 ± 5 | 12 ± 4 | ||||||
| TMB | TOL | OXYL | OCT | HONO | NO | NO2 | SO2 | RH | |
| 20110614a | 27 ± 6 | 128 ± 26 | 45 ± 9 | 22 ± 5 | 101 ± 6 | 85 ± 5 | 588 ± | 40 - 44 | |
| 168 | |||||||||
| BENZ | ISO | APIN | LIMO | ||||||
| 28 ± 6 | 4 ± 1 | 15 ± 3 | 17 ± 4 | ||||||
Temperature (K) recorded during the experiments.
| Hour | 20090610 | 20090622 | 20090623 | 20090624 | 20091005 | 20091006 |
|---|---|---|---|---|---|---|
| 7 | 293.3 | 296.5 | 295.8 | 294.4 | 291.8 | 291.4 |
| 8 | 296.1 | 297.6 | 297.8 | 296.8 | 293.3 | 292.7 |
| 9 | 299 | 300 | 299.3 | 299.6 | 296.2 | 295.2 |
| 10 | 300.1 | 301.3 | 299.8 | 301.8 | 299.1 | 298 |
| 11 | 310.1 | 302.8 | 301 | 305.4 | 302.4 | 297.6 |
| 12 | 313.1 | 303.2 | 304.1 | 306.7 | 303.6 | 298.9 |
| 13 | 314.2 | 304.6 | 305.7 | 307.1 | 304.6 | 302.9 |
| 14 | 312.8 | 304.5 | 306.6 | 306.8 | 300.4 | 303.3 |
| 15 | 308 | 302.3 | 305.1 | 306.8 | 300.4 | 302.8 |
| 16 | 300.4 | 303.5 | 303.2 | 302.8 | 300.4 | 300.1 |
| 17 | 298.7 | 301.9 | 300.5 | 302.8 | 300.4 | 297.8 |
| Hour | 20091007 | 20091008 | 20091014 | 20091026 | 20091027 | 20091029 |
|---|---|---|---|---|---|---|
| 7 | 292.3 | 293.5 | 291.2 | 293.4 | 289.8 | 290.1 |
| 8 | 293 | 294.5 | 292.1 | 293.9 | 290.3 | 290.3 |
| 9 | 294.5 | 296.7 | 293.3 | 295.8 | 292.4 | 292.1 |
| 10 | 295.6 | 299.6 | 296.9 | 297 | 295.1 | 293.9 |
| 11 | 298.9 | 302.1 | 300 | 299.8 | 297.8 | 297.7 |
| 12 | 298.2 | 304.9 | 301.9 | 301.5 | 300.2 | 301.8 |
| 13 | 300.9 | 302.5 | 301.6 | 302.1 | 301.1 | 302 |
| 14 | 300.2 | 300.8 | 301.4 | 301.9 | 300.7 | 301.1 |
| 15 | 299.1 | 304.1 | 300.8 | 300.7 | 298.7 | 297.2 |
| 16 | 295.5 | 301.8 | 298.6 | 297.4 | 294.6 | 295 |
| 17 | 295.5 | 301.8 | 298.6 | 297.4 | 294.8 | 293.9 |
| Hour | 20091030 | 20091103 | 20091106 | 20091111 | 20091112 | 20091116 |
|---|---|---|---|---|---|---|
| 7 | 291.1 | 286.5 | 287.2 | 288.5 | 288 | 290.7 |
| 8 | 291.8 | 286.6 | 287.4 | 288.8 | 288 | 291.1 |
| 9 | 294.1 | 288.8 | 288.4 | 290.4 | 289.6 | 292.8 |
| 10 | 295.4 | 291 | 290.6 | 292 | 291.7 | 295.8 |
| 11 | 298.6 | 292.4 | 295.2 | 295.3 | 295.4 | 298.5 |
| 12 | 300.9 | 297 | 295.1 | 298.1 | 298.6 | 301 |
| 13 | 302.1 | 299.7 | 294.3 | 299.7 | 298.5 | 302.6 |
| 14 | 301.6 | 301.7 | 295.5 | 299.7 | 296.7 | 302.6 |
| 15 | 299.7 | 302.6 | 294.5 | 298.3 | 296 | 301.1 |
| 16 | 295.8 | 300.6 | 291.6 | 296.1 | 292.1 | 296.5 |
| 17 | 295.8 | 296.1 | 291.6 | 293.4 | 291.1 | 294.4 |
| Hour | 20100607 | 20100608 | 20110614 | 20110616 | 20110617 | 20110621 |
|---|---|---|---|---|---|---|
| 7 | 295.6 | 293.7 | 292.8 | 295.3 | 296.2 | 295.5 |
| 8 | 295.6 | 293.7 | 293.9 | 295.1 | 297.6 | 297.5 |
| 9 | 297.8 | 294.9 | 295.4 | 299.3 | 299.7 | 300.3 |
| 10 | 299.8 | 297.3 | 297.2 | 301.9 | 302.5 | 302.2 |
| 11 | 301.9 | 299.1 | 302.5 | 304.3 | 306.1 | 305.2 |
| 12 | 302.3 | 301.7 | 305.4 | 305.2 | 307.5 | 305.2 |
| 13 | 303.5 | 304.6 | 306.5 | 306 | 307.5 | 305.2 |
| 14 | 305.2 | 306.2 | 306.4 | 306.1 | 307.4 | 305.2 |
| 15 | 305.3 | 306.6 | 305.2 | 306.3 | 307.4 | 305.2 |
| 16 | 300.8 | 306.8 | 303.2 | 302.9 | 307.4 | 305.2 |
| 17 | 300.8 | 306.8 | 299.5 | 299.2 | 307.4 | 305.2 |
A tapered element oscillating monitor (TEOM 1400a, Rupprecht & Patashnick) and a scanning mobility particle sizer (SMPS) with a particle counter (TSI 3022A CPC) and a differential mobility analyzer (TSI 3081 DMA), were used to study the aerosol particles formed during the experiments. These techniques were described in Vivanco et al. (2011).
For the monitoring of the gas phase, a gas chromatograph (GC), a gas chromatograph coupled to a mass spectrometer (GCMS) and an infrared spectrometer (FTIR) were used, together with several monitors.
Finally, considering that soluble sulphates and nitrates are the most relevant inorganic compounds, some filters were analyzed to discount the inorganic aerosol contribution using ion chromatography (Vivanco et al., 2011). Ammonium (NH4+) content was also determined in these experiments with a Dionex DX-500.
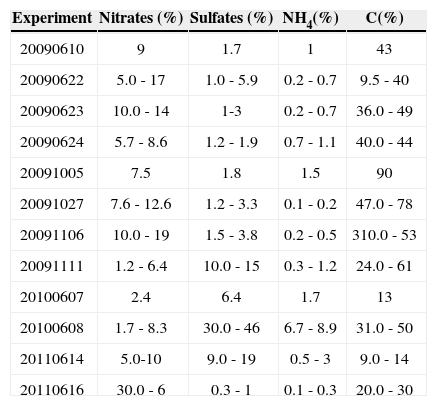
3Results3.1Inorganic contentResults for the ion chromatography elemental combustion analysis are summarized in Table III. The nitrate quantities detected by ion chromatography are in the same range as those presented in Vivanco et al. (2011). Again, sulfate content was negligible in the experiments with no SO2 (20090623, 20091027 and 20091106). In the experiments with initial SO2 (20091111, 20091112, 20100607 and 20100608), the sulfate content was within the range of 10-30%. The content of NH4+ was almost zero in all the experiments. Regarding the elemental analysis of carbon, the low carbon content found in some experiments, such as 20091106, seems to suggest the presence of highly oxidized organic compounds in the SOA phase.
Inorganic characterization (expressed as percentage) of the filtered aerosol mass collected in some experiments.a
| Experiment | Nitrates (%) | Sulfates (%) | NH4(%) | C(%) |
|---|---|---|---|---|
| 20090610 | 9 | 1.7 | 1 | 43 |
| 20090622 | 5.0 - 17 | 1.0 - 5.9 | 0.2 - 0.7 | 9.5 - 40 |
| 20090623 | 10.0 - 14 | 1-3 | 0.2 - 0.7 | 36.0 - 49 |
| 20090624 | 5.7 - 8.6 | 1.2 - 1.9 | 0.7 - 1.1 | 40.0 - 44 |
| 20091005 | 7.5 | 1.8 | 1.5 | 90 |
| 20091027 | 7.6 - 12.6 | 1.2 - 3.3 | 0.1 - 0.2 | 47.0 - 78 |
| 20091106 | 10.0 - 19 | 1.5 - 3.8 | 0.2 - 0.5 | 310.0 - 53 |
| 20091111 | 1.2 - 6.4 | 10.0 - 15 | 0.3 - 1.2 | 24.0 - 61 |
| 20100607 | 2.4 | 6.4 | 1.7 | 13 |
| 20100608 | 1.7 - 8.3 | 30.0 - 46 | 6.7 - 8.9 | 31.0 - 50 |
| 20110614 | 5.0-10 | 9.0 - 19 | 0.5 - 3 | 9.0 - 14 |
| 20110616 | 30.0 - 6 | 0.3 - 1 | 0.1 - 0.3 | 20.0 - 30 |
In order to estimate aerosol mass concentration from SMPS measurements, an aerosol density is required. We considered density values of 1.0 and 1.5g/cm3, in order to account for the range of density values reported by previous studies (Alfarra et al., 2006; Ng et al., 2007). Dilution and wall-losses corrections were calculated by considering the particle loss as a first order rate process. For that purpose, the concentration decay after the closure of the chamber was calculated, assuming that, once the chamber is closed, no more aerosols were formed and, therefore, the observed decay of the mass and volumetric concentrations can be attributed to particle losses (dilution and mainly wall deposition).
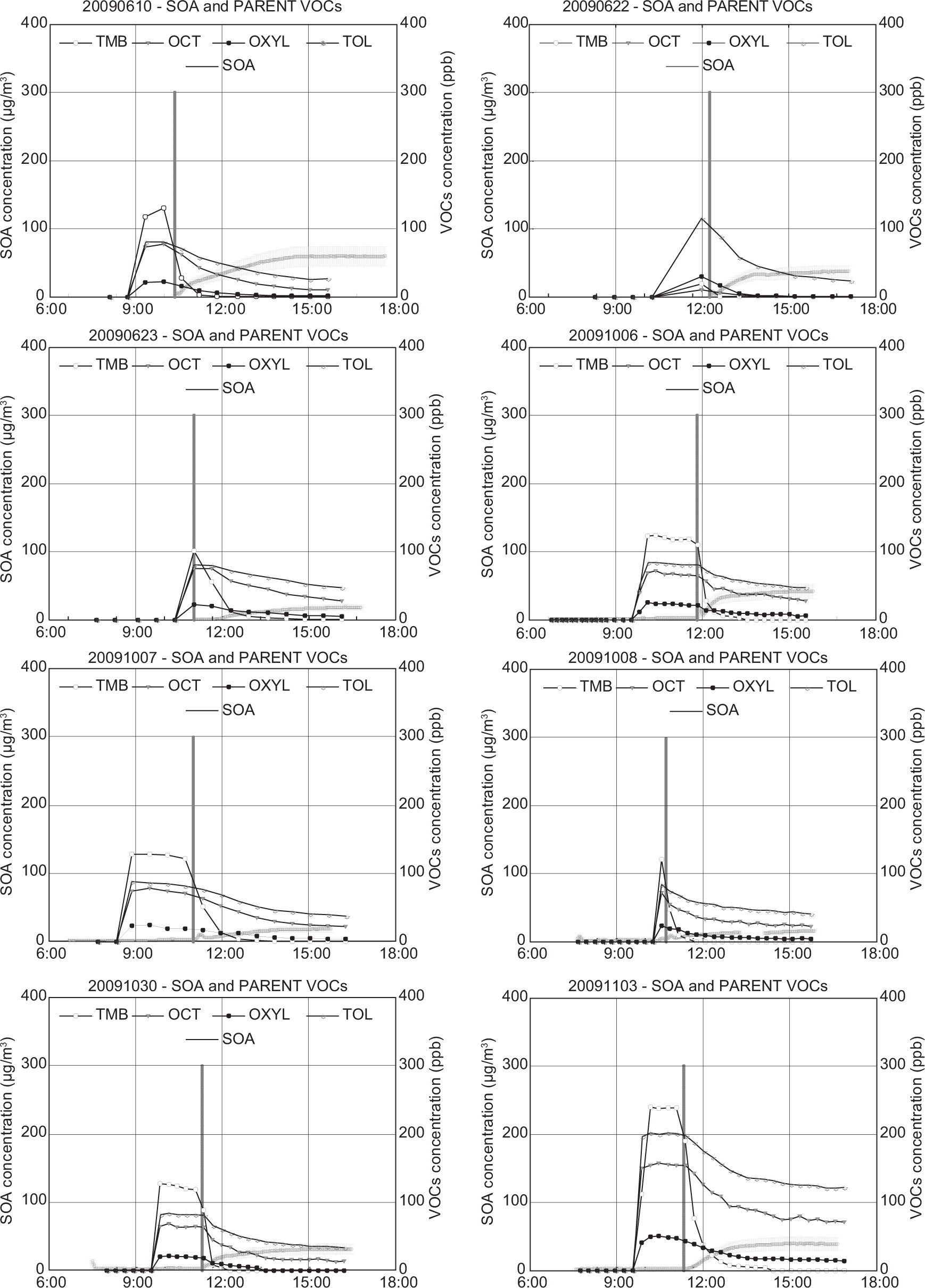
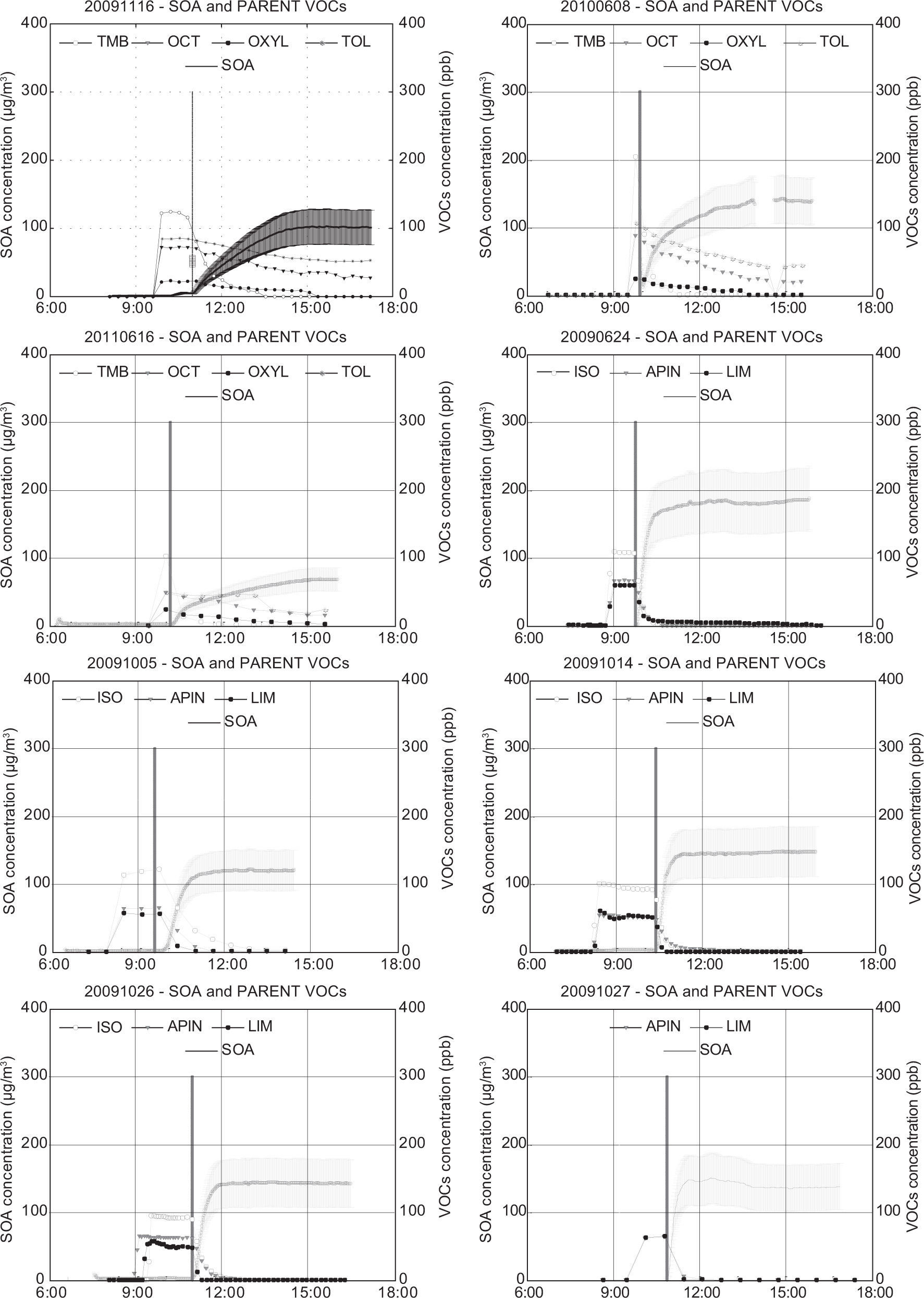
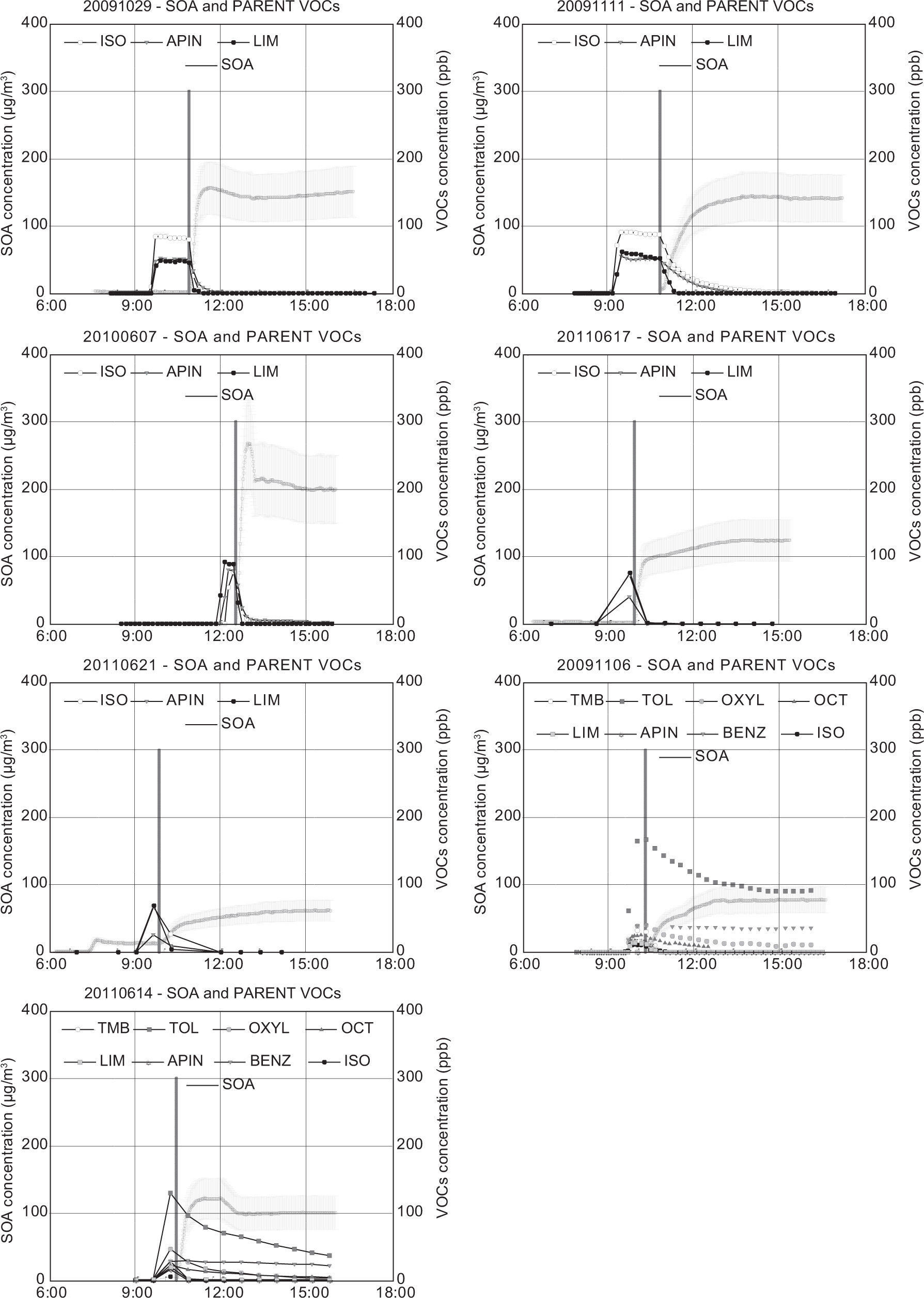
The inorganic fraction shown in Table III was discounted from the total SMPS aerosol and the remainder was assumed to be the SOA mass concentration. This assignment is neglecting aerosol water. Time series showing SOA concentration for each experiment are presented in Figures 1-3. These figures include the initial VOCs decay. The SOA mass concentration is presented as a range, with the upper and lower limits assuming an aerosol density of 1.0g/cm3 and 1.5g/cm3, respectively. The vertical black line indicates the opening of the chamber. The final SOA mass concentration and the SOA time profile during each experiment represent an important dataset for the evaluation of air quality model performance.
.")
.")
.")
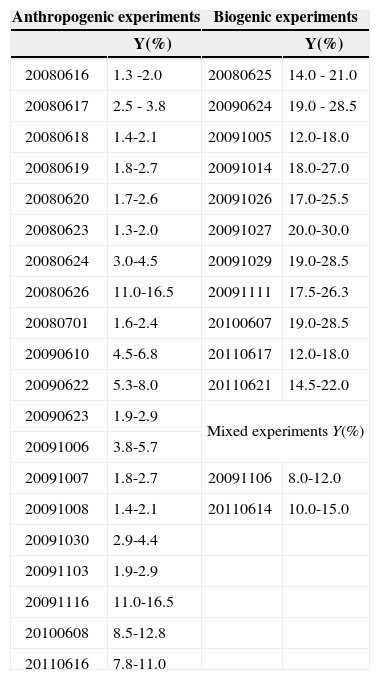
Aerosol yields (Table IV) were calculated following the procedure presented in Vivanco et al. (2011), but including wall-loss corrections. Since the yield values presented in Vivanco et al. (2011) were not corrected for losses, they were also recalculated here, though only small differences were found. SOA formation yields (Y) from the total mixture were estimated as the fraction of the reactive organic gases (ROG) that was converted to organic aerosol (Mo). This approach is usually applied to individual compounds, according to the gas/particle partitioning theory (Odum et al., 1996):
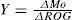
SOA yields (considering SMPS aerosol concentration) for the whole set of experiments. The ranges for Y are calculated with an upper and lower limit of 1.0 and 1.5g/cm3, respectively, for the SOA density.
| Anthropogenic experiments | Biogenic experiments | ||
|---|---|---|---|
| Y(%) | Y(%) | ||
| 20080616 | 1.3 -2.0 | 20080625 | 14.0 - 21.0 |
| 20080617 | 2.5 - 3.8 | 20090624 | 19.0 - 28.5 |
| 20080618 | 1.4-2.1 | 20091005 | 12.0-18.0 |
| 20080619 | 1.8-2.7 | 20091014 | 18.0-27.0 |
| 20080620 | 1.7-2.6 | 20091026 | 17.0-25.5 |
| 20080623 | 1.3-2.0 | 20091027 | 20.0-30.0 |
| 20080624 | 3.0-4.5 | 20091029 | 19.0-28.5 |
| 20080626 | 11.0-16.5 | 20091111 | 17.5-26.3 |
| 20080701 | 1.6-2.4 | 20100607 | 19.0-28.5 |
| 20090610 | 4.5-6.8 | 20110617 | 12.0-18.0 |
| 20090622 | 5.3-8.0 | 20110621 | 14.5-22.0 |
| 20090623 | 1.9-2.9 | Mixed experiments Y(%) | |
| 20091006 | 3.8-5.7 | ||
| 20091007 | 1.8-2.7 | 20091106 | 8.0-12.0 |
| 20091008 | 1.4-2.1 | 20110614 | 10.0-15.0 |
| 20091030 | 2.9-4.4 | ||
| 20091103 | 1.9-2.9 | ||
| 20091116 | 11.0-16.5 | ||
| 20100608 | 8.5-12.8 | ||
| 20110616 | 7.8-11.0 | ||
To estimate the “total” yields we considered the total organic gas reacted when maximum aerosol concentration was formed, by adding together the single VOCs mass reacted.
Most of the anthropogenic experiments present SOA yields in the range of 1-7%. SOA formation in these experiments is mainly governed by the photooxidation of 1,3,5-TMB, due to the high initial concentrations introduced in each experiment compared to the rest of VOCs. Substituted aromatics such as 1,3,5-TMB and o-xylene are commonly referred to as low-yield aromatics, due to their lower yield compared to high-yield aromatics such as toluene or benzene. Odum et al. (1997) reported yields within the range of 3-5 % and 7-10 % for the low-yield and high-yield aromatics, which have been confirmed by other studies (Cocker et al., 2001; Ng et al, 2007; Healy et al., 2008). These values agree with the yields obtained in most of our anthropogenic experiments. A greater SOA yield was found for the experiment 20090622, in which a larger amount of toluene was introduced into the chamber and therefore the SOA yield obtained was higher (5-8 %), due to the major contribution of high-yield aromatic toluene to the overall SOA yield. However, the highest SOA yields for the anthropogenic experiments were obtained in the experiments 20080626, 2009116, 20100608 and 20110616. In the experiment 20110616, the higher yield may be related to the elevated relative humidity employed, as some studies have detected a greater SOA formation from aromatic VOCs under high relative humidity conditions (Hu and Kamens, 2007). In the other three experiments, the higher values are related to the inclusion of SO2 in the chamber. During the last years, several authors have reported the existence of an important group of heterogeneous acid-catalyzed reactions, in which oxygenated compounds such as glyoxal, methylglyoxal or octanal formed through gas-phase oxidation of aromatic VOCs and alkanes, react in the presence of sulfuric acid particles to form low volatility products that increase the total SOA mass of the system (Jang and Kamens, 2001; Jang et al., 2002).
Regarding the biogenic experiments, higher yield values were obtained compared to the anthropogenic experiments (between 9 and 30%, considering all the experiments). Some authors have reported high monoterpenes SOA yields, with values up to 12 and 40% for a-pinene and limonene, respectively (Odum et al., 1996; Hoffmann et al., 1997; Leungsakul et al., 2005), in agreement with our results. On the other hand, isoprene is a VOC with a relatively low SOA yield (1-5%) and thus its presence in the mixture reduces the overall yield, as it is quickly consumed without a high formation of SOA (Dommen et al., 2006; Kroll et al., 2006). Because of that reason, experiment 20091027 presents the highest SOA yield, as no isoprene was included in the mixture. Although recent studies have pointed out the influence of SO2 in SOA formation from isoprene and α-pinene photooxidation (Surratt et al., 2007a, b), we have not observed a significant yield increase in the experiments in which initial SO2 was introduced in the chamber (20091111 and 20100607), likely due to variations in other experimental conditions.
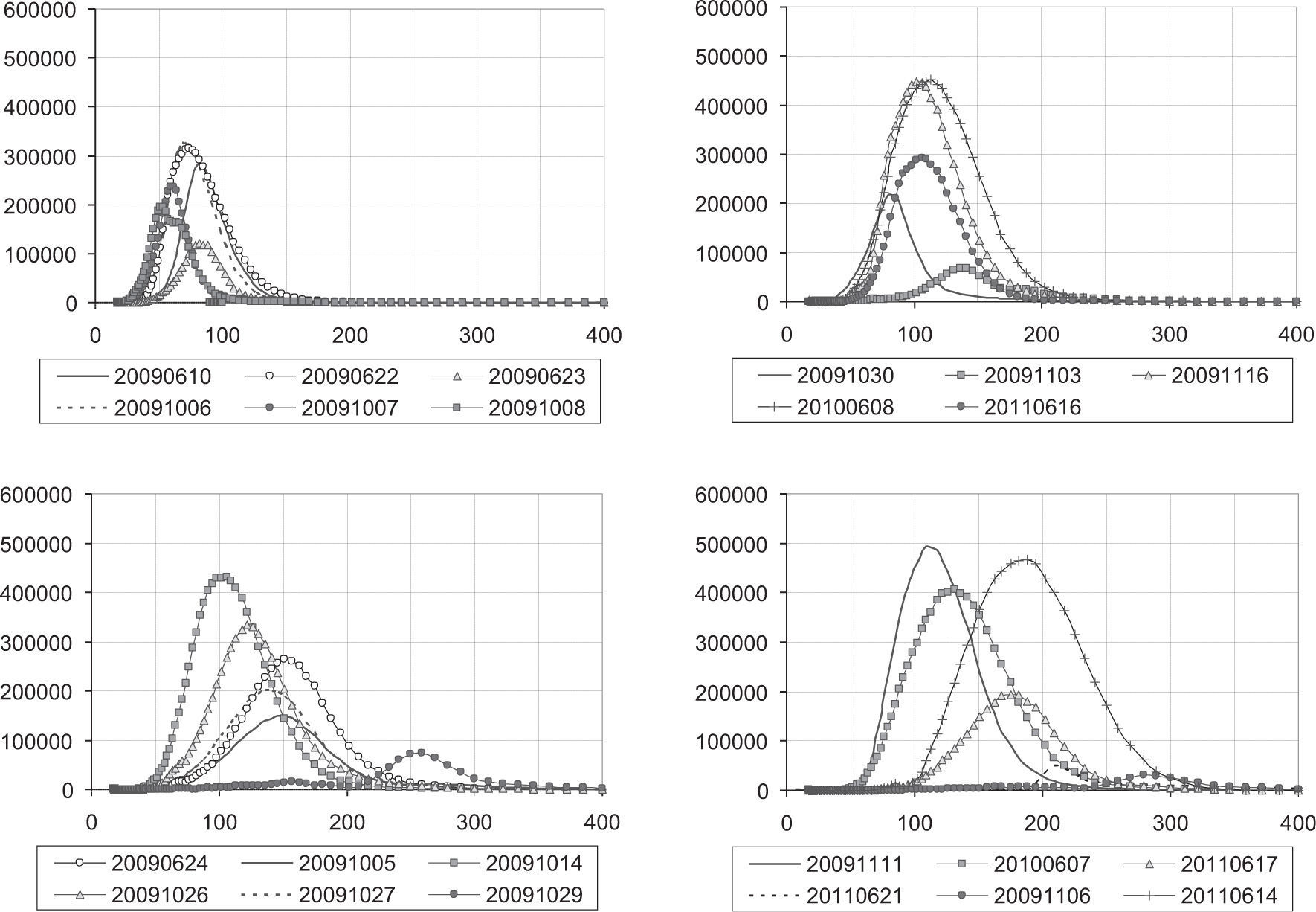
3.3Particle sizeFinally, particle size distributions measured two hours after the opening of the chamber are presented in Figure 4. Higher mean diameters were reached for the experiments with biogenic parent VOCs; for most of them the size distribution was centered on the range 100-150nm (20091014, 20091026, 20091111 and 20090607). Slightly coarser particles were found in experiments 20091005 and 20090624. The largest sizes among the biogenic mixtures were observed in experiment 20091029, with a particle size distribution function centered on approximately 250nm. In the anthropogenic experiments, almost all the experiments present a mean diameter in the range of 50-100nm, with the exception of the experiments with SO2, which present the coarsest particles, and the experiment with 70% of relative humidity (20110616). In the mixed experiments, the coarsest particles were observed in the experiment 20091106, although this behavior can be also related to the dry conditions used. Experiment 20110614 also generated large particles, in this case with an important number of particles due to the presence of sulphates in the experiment. Note that although concentration values in Figures 1-3 were corrected for inorganic aerosols, values in Figure 4 are not.
4Conclusions while X-axis represents mean particle diameter (nm).")
New experiments performed in 2009, 2010 and 2011 in the EUPHORE chamber supplement the experimental data presented in Vivanco et al. (2011). In general, SOA yields and particle sizes were higher in the experiments with biogenic VOCs, compared to the experiments with anthropogenic VOCs. The presence of SO2 increased SOA yields, providing additional evidence of the SOA formation enhancement in acidic conditions previously mentioned in Vivanco et al. (2011) for the anthropogenic mixtures. This behavior was not so clearly observed for the biogenic experiments. Also coarser particles were observed in the presence of initial SO2.
This set of data provides a relevant support for model performance evaluation activities, as measurements of explicit SOA concentration are not generally available (and model community must currently apply certain approaches to evaluate model results). With these experiments the direct comparison between models and observation is feasible. Even the evaluation of specific processes, such as aerosol and chemistry modules, can be done, as no other processes due to transport and mixing are involved inside the chamber. When dealing with air quality measurements, the evaluation of these modules cannot be done directly, as other atmospheric processes affect SOA concentrations.