


Introducción: El síndrome de DRESS (Drug Rash with Eosinophilia and Systemic Symptoms) se presenta por una reacción adversa grave a medicamentos. Usualmente se define por la triada de fiebre, exantema y afectación sintomática o asintomática de órganos internos. Los criterios diagnósticos son la sospecha de una reacción medicamentosa, eosinofilia (≥ 1.5×109/l y/o linfocitos atípicos en sangre periférica) y afectación de dos o más órganos internos, incluida la piel. La incidencia estimada de este síndrome varía de 1/1000 a 1/10,000 de los pacientes expuestosa medicamentos. Presenta una mortalidad hasta del 30%.
Caso clínico: Adolescente femenino de 14 años con historia de crisis convulsivas parciales complejas secundarias a trauma de cráneo, quien inició tratamiento con carbamazepina. Después de cuatro semanas presentó fiebre, exantema generalizado, adenopatías y compromiso multisistémico. Después de estudios paraclínicos y valoración por diversos especialistas, se estableció el diagnóstico de síndrome de DRESS. Se suspendió el tratamiento con carbamazepina, y se administraron esteroides y gammaglobulina. Se observó buena respuesta y la remisión de lasintomatología.
Conclusiones: La suspensión inmediata del fármaco causante del síndrome y la iniciación del tratamiento con corticosteroides sistémicos son los pilares en el tratamiento.
Background: DRESS syndrome (Drug Rash with Eosinophilia and Systemic Symptoms) is a serious adverse reaction to medication. It is usually defined by the triad of fever, rash and symptomatic or asymptomatic involvement of internal organs. Diagnostic criteria are suspected drug reaction, eosinophilia (≥1.5 ×109/l and/or atypical lymphocytes in peripheral blood) and involvement of two or more internal organs (including the skin). The estimated incidence of this syndrome ranges from 1/1000 to 1/10,000 drug exposures and up to 30% mortality.
Case report: We present a 14-year-old female with a history of complex partial seizures secondary to head trauma. She began treatment with carbamazepine. After 4 weeks she developed fever, generalized rash, adenopathy and multisystem involvement. Following paraclinical studies and evaluation by various specialists, DRESS Syndrome diagnosis was established. The patient was treated with carbamazepine suspension, steroids and gammaglobulin administration with good response and remission of symptoms.
Conclusions: Immediate withdrawal of the causative drug and initiation of systemic corticosteroids is the mainstay in disease management.
Pagina nueva 1. Introduction
DRESS syndrome (Drug Reaction (or Rash) with Eosinophilia and Systemic Symptoms) is caused by an adverse reaction to medications. This is a rare, severe and multiorgan reaction found frequently associated with antiepileptic agents (phenytoin, carbamazepine, phenobarbital, lamotrigine) allopurinol, dapsone and sulfonamides.1,2
WHO defines adverse drug reactions to medications as an unintended response to a noxious drug and which occurs when administered at normally used doses in humans for prophylaxis, diagnosis or disease treatment or for modification of a physiological function.3
DRESS syndrome is distinguished by the clinical triad of fever, rash and multiorgan involvement.4 The pathology is partially known as different mechanisms have been implicated in its development.5 It has been reported that there could be an immunological phenomenon involved in its origin that causes the release of cytokines by the T-lymphocytes and activation of macrophages.6 Other mechanisms include defects in detoxification that lead to the formation of reactive metabolites and subsequent immunological reactions, of slow acetylation and reactivation of human herpes, including types 6 and 7 (HHV-6 and HHV-7), and of the Epstein-Barr virus.7 DRESS syndrome is of late beginning and with slow evolution and is clinically similar to infectious processes, for which it is frequently misdiagnosed.8 Its incidence varies from 1/1,000-1/10,000 of individuals exposed to different drugs.5 Correct diagnosis is difficult due to the variety of clinical and laboratory abnormalities. Incorrect diagnosis or diagnostic delay has translated to a mortality increase of up to 30%.2,7 Immediate suspension of the culprit drug and start of treatment with systemic corticosteroids is the cornerstone of treatment for DRESS syndrome.9 We present the case of a patient with difficulty of establishing a diagnosis due to the evolution of the symptoms observed. However, once correctly diagnosed, the patient had a satisfactory evolution.
2. Clinical case
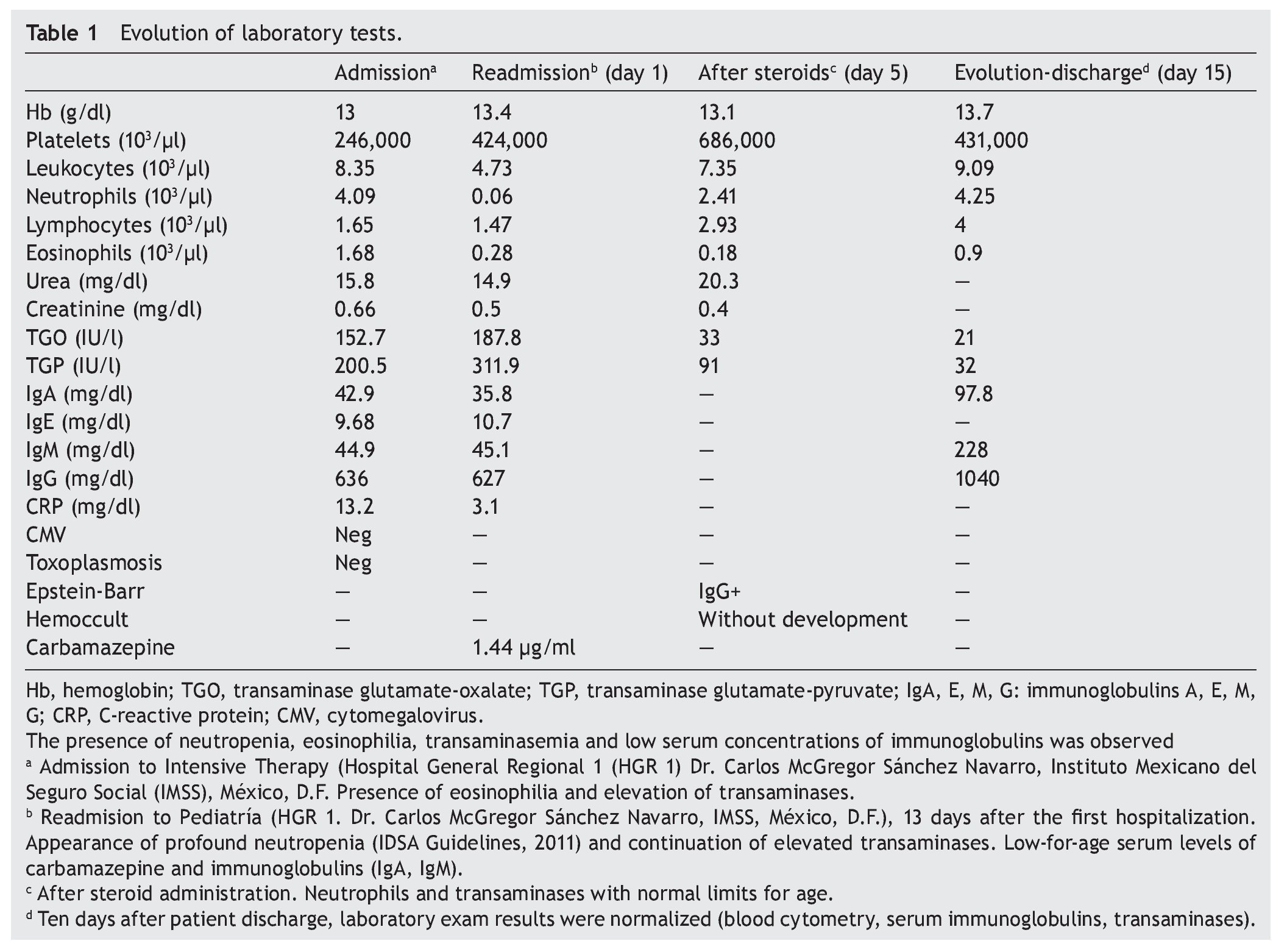
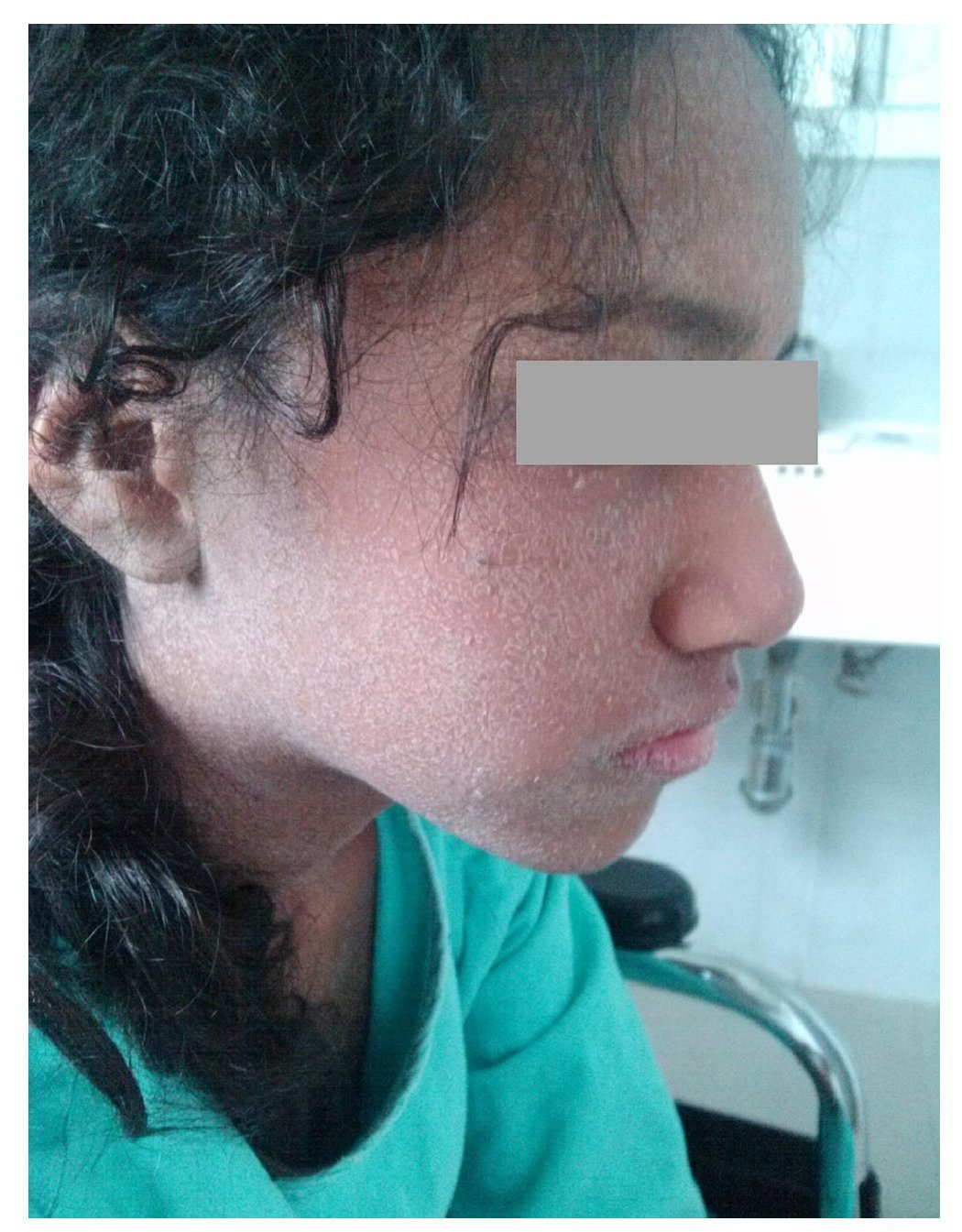
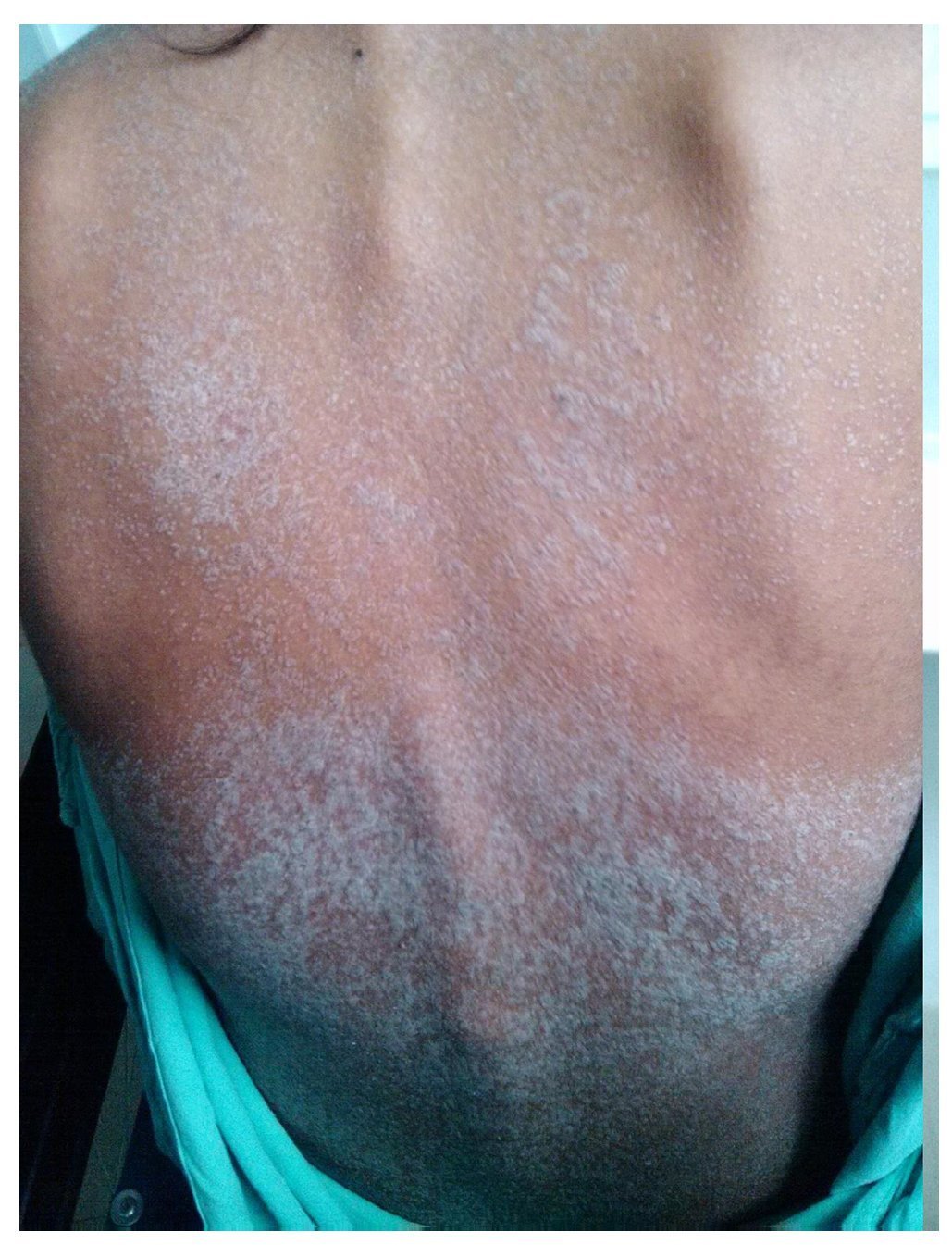
We present the case of a 14-year-old female patient with percentiles from 10-25 for weight, height and BMI for age. The patient has a history of partial complex seizures of 3 months evolution secondary to head trauma due to a fall. Plain and contrast computed tomography (CT) of the skull showed no structural changes. EEG showed a slow wave point complex that begins in the right temporal region, which is subsequently repeated during the sleep phase. Treatment was started with carbamazepine (9 mg/kg/day). During the fifth week she had a clinical picture characterized by fever, odynophagia, maculopapular rash with fine desquamation starting in the face, with cephalocaudal progression, pruritis and facial involvement characterized by edema and erythema (Fig. 1). Treatment was based on β-lactams. Due to the worsening symptoms, she was diagnosed with severe scarlet fever vs. Stevens-Johnson Syndrome and was admitted to the intensive care. She had a discrete improvement of symptoms; therefore, it was decided to complete β-lactam treatment for 7 days at home. She then started once again with fever and maculopapular eruption with fine desquamation in the face, trunk and extremities (Fig. 2) as well as hepatic pain. She was readmitted to the hospital and, due to the clinical evolution and analysis of paraclinical studies (Table 1), diagnosis of DRESS syndrome was considered. She was placed on isolation, carbamazepine was stopped and serum levels were monitored. Temperature was controlled along with hyper-hydration, fasting, clindamycin, methimazole, hydroxyzine and methylprednisone (30/mg/kg single dose), with almost total symptom remission in the first 8 h. She was evaluated by the hematology staff who broadened the antimicrobial therapy adding amikacin and fluconazole due to severe neutropenia and fever. Cardiology documented a structurally healthy heart. Evaluation by dermatology resulted in adjustment of the hydroxyzine dose. On the fourth day she was given a single dose of IV immunoglobulin G (600 mg/ kg) and prednisone was started (1 mg/kg/day). On the 7th day she had complete remission of the skin lesions (Fig. 3). She completed antimicrobial therapy and was discharged with prednisone in tapering doses and begun on levetiracetam, although she still had mild alterations in liver enzymes, which were completely normalized 15 days later (Table 1).
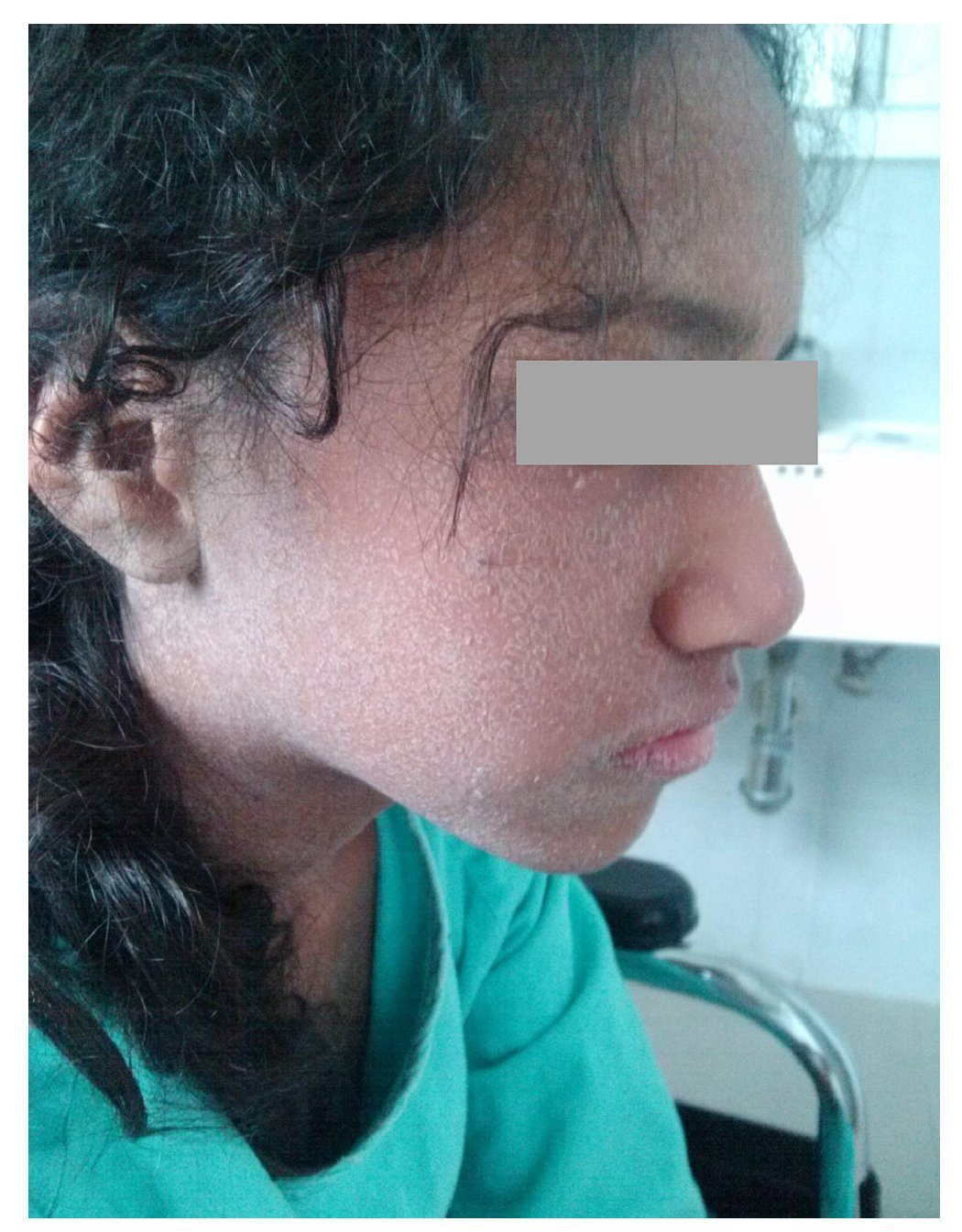
Figure 1 Lesions observed at the time of the diagnosis of DRESS syndrome characterized by generalized fine desquamation.
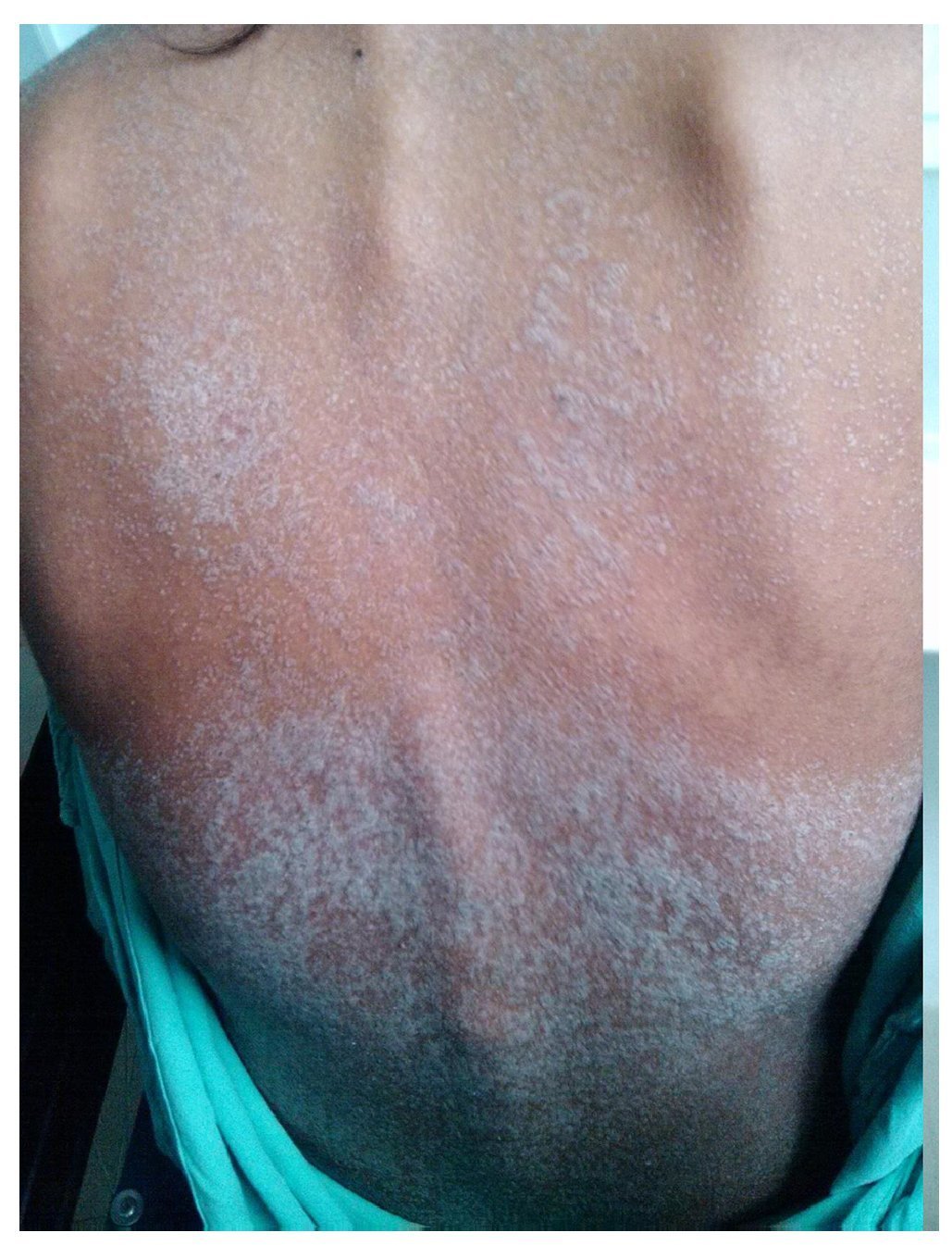
Figure 2 Generalized dermatosis consisting of scaly plaques predominantly on the trunk and extremities.
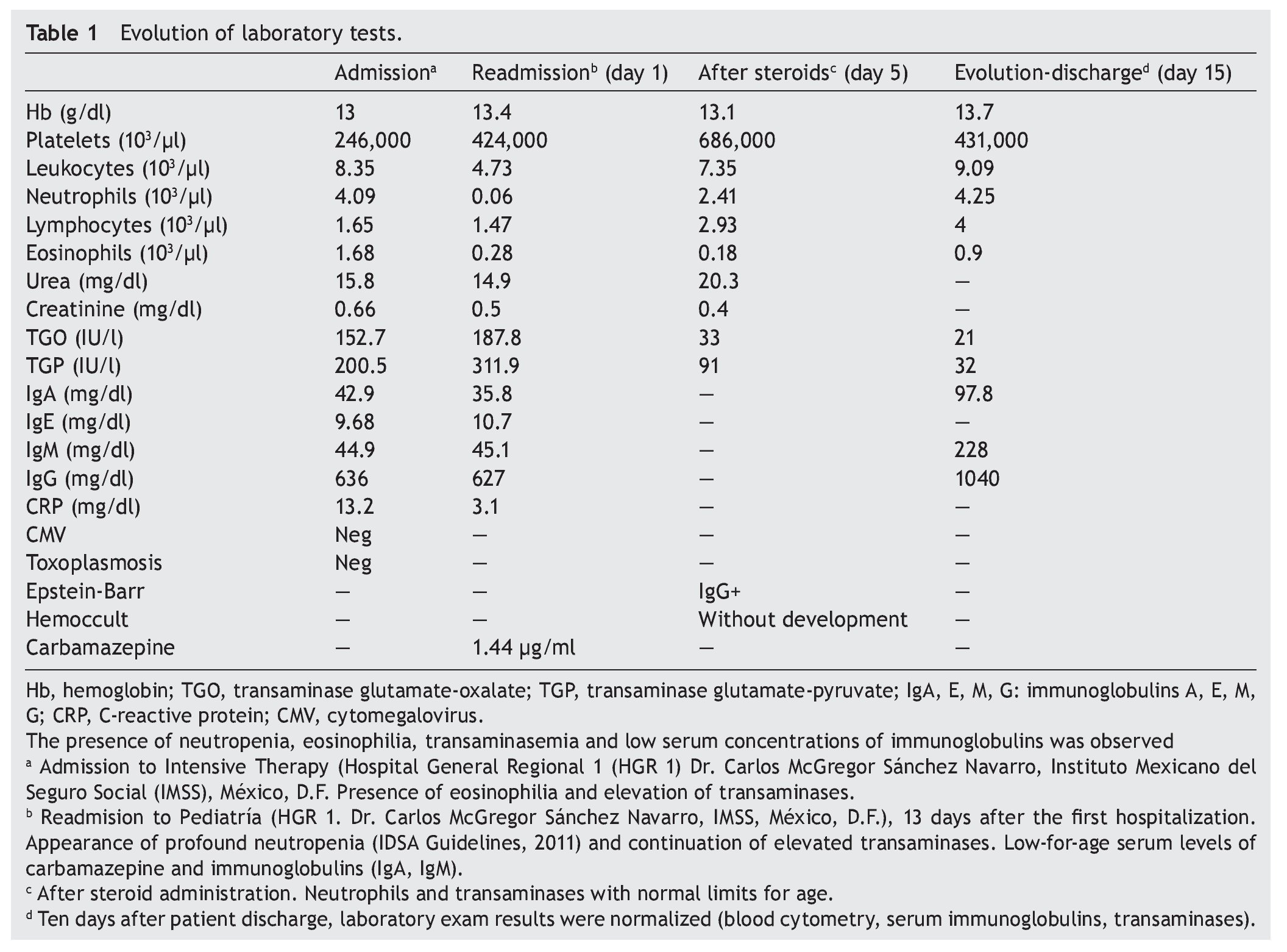
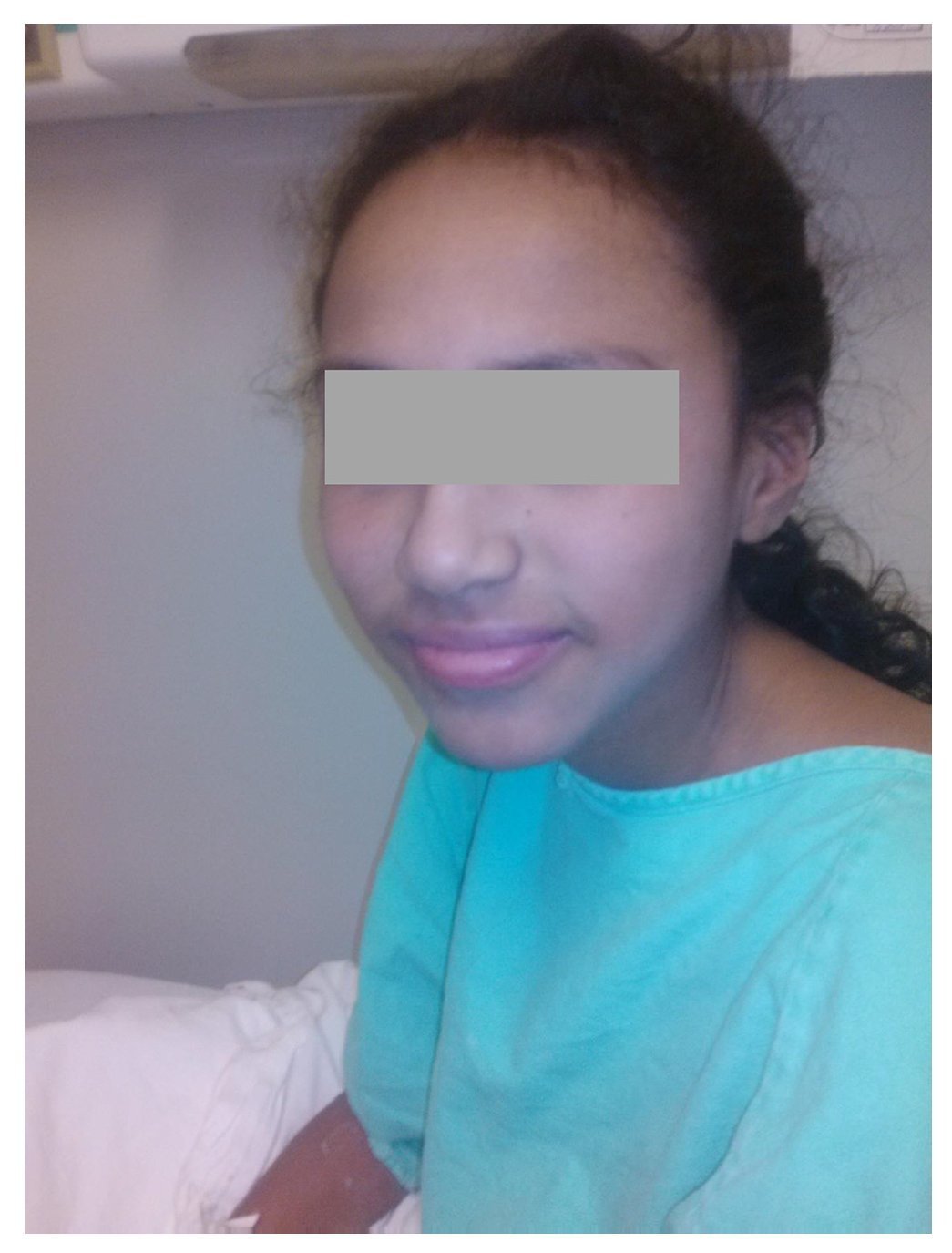
Figure 3 Patient at time of discharge after administration of intravenous steroids, with disappearance of scaly lesions.
3. Discussion
Adverse skin drug reactions affect ~2-3% of hospitalized patients who receive some type of systemic treatment.10 Aromatic anticonvulsant drugs, antibiotics and nonsteroidal anti-inflammatory drugs are not drugs that are commonly associated with this clinical condition.10,11 Pediatric patients are at greater risk of developing this condition due to the greater incidence of seizures in the first decade of life, as well as slow acetylators and patients with in vitro hypersensitivity to hydroxylamine metabolites.10
Hypersensitivity reactions to carbamazepine are type B reactions. These cannot be predicted, do not show an obvious relationship to the dosage and affect a minority of patients, which suggests it is dependent on factors inherent to the host and cannot be reproduced in animal models.12
DRESS syndrome is a severe adverse reaction to medication associated with the use of aromatic anticonvulsants and allopurinol.13 In 1996, Bocquet et al. proposed the acronym DRESS (Drug Reaction (or Rash) with Eosinophilia and Systemic Symptoms) to describe a potentially fatal syndrome that includes a severe skin eruption, fever, hematological alterations (eosinophilia or atypical lymphocytes) and involvement of internal organs.14 The estimated incidence of this syndrome varies from 1/1,000-1/10,000 of patients exposed to the medication.5 However, the term DRESS has been inconsistent because eosinophilia is not a constant finding and the dermatological and systemic signs are variable.15
Family history of DRESS syndrome, the presence of primary or acquired immunodeficiencies, and neoplasms are the most common risk factors for developing this type of skin drug reaction.16 The disease is partially known. Different mechanisms have been implicated in its development including immunological factors, genetic factors (the risk increases up to 25% if a first degree relative has this syndrome), defects of detoxification that lead to the formation of reactive metabolites and subsequent immunological reactions, slow acetylation and reactivation of human herpes, including the HHV-6 and HHV-7, and the Epstein-Barr virus. Detection of HHV-6 reactivation has been recently proposed as a diagnostic marker for DRESS.5,7,17
In a study carried out by McCormack et al., it was found that one variation in the presence of the HLA-A*3101 allele is an important predictor of entire spectrum of reactions of hypersensitivity to carbamazepine in persons with European ancestry. Although the presence of the HLA-A*3101 allele is not necessary or sufficient for developing carbamazepine hypersensitivity, it is associated with a significantly greater risk. Persons with hypersensitivity syndrome caused by carbamazepine have demonstrated to have various types of T-cells specific for the drug in peripheral blood, including CD4+, CD8+ and CD4-CD8+ cells.18
Clinical manifestations of DRESS syndrome tend to appear at 2 to 8 weeks of exposure to the drug or within the first hours if there is prior sensitivity.10,19 Fever is frequently the first symptom. Skin lesions (present in 85% of cases) appear 24 to 48 h later. They consist of erythematous morbilliform macular eruptions localized on the face, trunk and extremities that become papular erythematous eruptions, which are confluent, symmetrical and pruritic and with edematous and especially perifollicular infiltration. Facial edema, mainly on the forehead, periorbital region, hands and feet, as well as the presence of conjunctivitis is highlighted. This eruption could be associated with vesicles, pustules, atypical target lesions, purpura and desquamation.20 Involvement of the mucosa is uncommon although it could manifest as conjunctivitis, chelitis, and mouth and genital erosions. Facial edema can be disfiguring for the patient.
Among the extracutaneous manifestations, patients present with bilateral and symmetrical adenopathies >2 cm in diameter and also have hepatosplenomegaly.10 The systemic compromise tends to begin at 1 to 2 weeks after the skin eruption, with liver involvement being the most common and the principal cause of death. There may be asymptomatic hepatomegaly, hepatitis with mild increase in transaminases or fulminant hepatitis.16,20 The kidney is secondarily involved with interstitial nephritis; however, in general, any organ could be involved.20
Involvement of the lung is uncommon and may be characterized by dyspnea, dry cough, bronchospasm, hypoxemia and bilateral pulmonary infiltrates. Cases of pneumonitis, bronchiolitis obliterans and pneumonia have been described.10
In terms of cardiac involvement, it may manifest as carditis or pericarditis. Myocarditis occasionally presents itself weeks or months after the DRESS episode. The patient manifests chest pain, dyspnea, tachycardia and hypotension with cardiomegaly, pleural effusion and arrhythmias; sometimes it may occur several weeks or months after cessation of the medication.
Of the hematological disorders, anemia and lymphocytosis are notable (which can present atypical lymphocytes and eosinophilia in 70% of the cases). These are the most common. Agranulocytosis, aplastic or hemolytic anemia and/or thrombocytopenia and hypoalbuminemia may also be present.10
Other nonspecific manifestations are arthritis, myositis, pancreatitis, meningoencephalitis, thyroid disorders (hypothyroidism, thyroiditis), syndrome of inappropriate antidiuretic hormone secretion, orchitis and parotiditis.10
Diagnosis of this problem may be complex. There are no established criteria. Diagnostic criteria have been proposed based on the cutaneous and hematological disorders and systemic involvement. These criteria, although without consensus, could be useful. Diagnostic criteria are as follows:
1. Suspected medication reaction
2. Hematological abnormalities (eosinophilia ≥1.5 × 109/l and/or atypical lymphocytes in peripheral blood)
3. Systemic compromise: adenopathies >2 cm in diameter, hepatitis (transaminases greater than double the normal value) and interstitial nephritis.
4. Presence of the three is necessary.6,10
According to the score by Kardaun et al., this patient was classified as definitively having DRESS syndrome by having 6 points.21 The present case, as reported in the literature, required the intervention of different medical specialists as well as intensive care management. The symptoms began in the fifth week with subtherapeutic serum levels of carbamazepine. The significant therapeutic response to the suspension of carbamazepine plus the use of steroids, as well as the overall analysis of the clinical progression and paraclinical studies, confirmed the diagnosis.
Although the prevalence is low, it is important to consider this diagnosis because the misdiagnosed cases (due to lack of knowledge about the disease) eventually lead to death.
It is possible that the interrelation of constitutional and acquired factors alter the ability of the biotransformation of reactive metabolites of the medication, the active infection due to HHV-6 (reactivation or new infection), the production of anti-cytochrome P450 antibodies, and the development of specific T-cell response.22 This case showed IgG positivity for the Epstein-Barr virus.
One of the principal causes of death is the hepatic compromise, which is characterized by anicteric cholestasis that could evolve to frank hepatitis with functional alteration, at times progressive despite suspension of the offending medication. With respect to the management of these patients, a point worth considering is the fact that the first-degree relatives of an affected individual have a four times greater risk to sensitivity to the same drugs than the general population so that family counseling has great importance.23
The therapy for DRESS syndrome is a challenge. The most important measures are early recognition of the syndrome and immediate suspension of the presumed drug. Delay could be associated with poor results. Supportive therapy should be provided to stabilize the patient. It should include antipyretics to reduce the effects of fever and topical steroids to alleviate skin symptoms. Administration of empiric antibiotics or anti-inflammatory drugs is contraindicated during the acute phase of DRESS syndrome because the clinical condition could be confused or exacerbated as a result of an unexplained cross-reactivity between medications.9 In this case, the treatment team failed by indicating empiric antibiotic therapy and by not immediately ceasing any suspected drug.
When there is exfoliative dermatitis, therapy is similar to a large burn area and could be provided in a specialized intensive care unit or in a burn unit. The measures include fluid replacement, correction of electrolyte imbalance, moderate elevation of room temperature, high caloric ingestion, treatment of superinfections and bacteremias with antibiotics and skin care of with appropriate bandages.9
In general, early administration of systemic steroids for all cases of DRESS syndrome is recommended. Topical corticosteroids can be applied to the skin lesions for symptomatic relief. Treatment with systemic steroids should begin with a minimum dose of 1 mg/kg/day of prednisone or its equivalent. To avoid a relapse, gradual reduction of the drug from 3 to 6 months after clinical and laboratory stabilization is recommended.9 In cases in which there is no improvement, worsening of symptoms with oral corticosteroids or significant visceral involvement, the patients can be treated with intravenous methylprednisolone (boluses of 30 mg/kg IV) for 3 days.9 Patients with life-threatening signs (hemophagocytosis with bone marrow insufficiency, encephalitis, severe hepatitis, renal and respiratory insufficiency) could be treated with IV steroids and immunoglobulin at a dose of 2 g/kg for 5 days. Immunoglobulin should not be administered without associated steroids. In cases with signs of severity and the confirmation of the great viral reactivation, antiviral drugs such as ganciclovir can be given as well as steroids and immunoglobulin. It is important to provide long term follow-up with laboratory tests to control a relapse.9
In conclusion, DRESS syndrome is a rare, severe, potentially fatal reaction to medications; therefore, its early diagnosis is important. Usually it is defined by the clinical triad of fever, exanthema, and multiorgan involvement. Diagnosis is clinical and should be suspected in a patient who receives anticonvulsant medications or some other suspected medication added to clinical manifestations reported. Treatment is based on immediate suspension of the culprit medication and the initiation of systemic corticosteroids with which the morbidity and mortality could be decreased.
Ethical responsibilities
Protection of human and animal subjects. The authors declare that no experiments were performed on humans or animals for this investigation.
Confidentiality of Data. The authors declare that no patient data appear in this article.
Right to privacy and informed consent. The authors declare that no patient data appear in this article.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 14 October 2014;
accepted 17 March 2015
http://dx.doi.org/10.1016/j.bmhimx.2015.03.007
* Corresponding author.
E-mail:diaquimar@hotmail.com (D.C. Quintero-Martínez).