



La hormona antidiurética arginina vasopresina (AVP) es liberada de la hipófisis, y regula la reabsorción de agua en las células principales del túbulo colector renal. La unión de la AVP al receptor tipo 2 de la AVP en la membrana basolateral induce la translocación de los canales acuosos de la acuaporina-2 hacia la membrana apical de las células principales de los túbulos colectores, induciendo la permeabilidad al agua de la membrana. Lo anterior da como resultado la reabsorción de agua en el túbulo colector de la nefrona, bajo la influencia de un gradiente osmótico.
La diabetes insípida nefrogénica es causada por la resistencia parcial o total al efecto de la AVP. La diabetes insípida nefrogénica congénita es una alteración asociada con mutaciones en los genes AVPR2 o AQP2, ocasionando la incapacidad del paciente para concentrar la orina. La diabetes insípida nefrogénica adquirida o secundaria puede ser causada por desbalances electrolíticos (hipercalcemia, hipokalemia), enfermedades renales o extrarrenales y fármacos (toxicidad por litio).
En este artículo se revisan las causas, manifestaciones clínicas, diagnóstico y tratamiento de los pacientes con diabetes insípida nefrogénica. También, con base en la comprensión de los mecanismos íntimos de la alteración, se exploran nuevas estrategias terapéuticas.
The anti-diuretic hormone arginine-vasopressin (AVP) is released from the pituitary and regulates water reabsorption in the principal cells of the kidney collecting duct. Binding of AVP to the arginine-vasopressin receptor type-2 in the basolateral membrane leads to translocation of aquaporin-2 water channels to the apical membrane of the principal cells of the collecting duct, inducing water permeability of the membrane. This results in water reabsorption in the collecting duct of the nephron following an osmotic gradient.
Nephrogenic diabetes insipidus is caused by partial or complete renal resistance to the effects of AVP. Congenital nephrogenic diabetes insipidus is a disorder associated with mutations in either the AVPR2 or AQP2 gene, causing the inability of patients to concentrate their urine. Acquired nephrogenic diabetes insipidus can be caused by electrolyte imbalances (e.g., hypercalcemia, hypokalemia), renal/extra-renal diseases and drugs (e.g., lithium toxicity).
This article reviews the causes, clinical manifestations, diagnosis and treatment of patients with nephrogenic diabetes insipidus. Based on more in-depth mechanistic understanding, new therapeutic strategies are current being explored.
1. Introduction
Diabetes insipidus is a disease characterized by the elimination of high volumes of very dilute urine. This disorder is caused by the failure of the posterior pituitary to secrete adequate amounts of arginine vasopressin (AVP), also called antidiuretic hormone (neurogenic or central diabetes insipidus), or by the inability of the kidney to respond to circulating AVP (nephrogenic diabetes insipidus).1
AVP exercises important effects on the excretion of urine and, with it, on fluid equilibrium. It is a nine amino acid peptide with an annular structure and a disulfuric connection. AVP is synthesized by large body neurons (magnocellular) located in the supraoptic and paraventricular nuclei of the hypothalamus. Its synthesis is accompanied by the generation of a specific carrier protein called neurophysin II. AVP unites to neurophysin and as such is transported by the axons of the hypothalamus-pituitary fascicle at a rate of 2 or 3 mm/h up to the neurohypophysis where the hormonal complexes are stored in the shape of granules in the nerve endings until they are used. When adequate stimuli are received, the hormone is secreted together with neurophysin by means of exocytosis. The process of secretion requires calcium entry through the membrane. Once this occurs, the granule membrane is reshaped by the cell after the microvesicles are covered and formed.1,2
The most important biological action of AVP is preservation of body water by reducing urinary output. This antidiuretic effect is obtained by promoting water reabsorption in the collecting tubule of the nephron. AVP induces antidiuresis via its interaction with the V2 receptors of the AVP in the kidney. Increase in the permeability to water in the collecting tubule of the nephron implies action of the aquaporin-2 water channel in the apical membranes of the principal cells of this segment of the renal tubule.
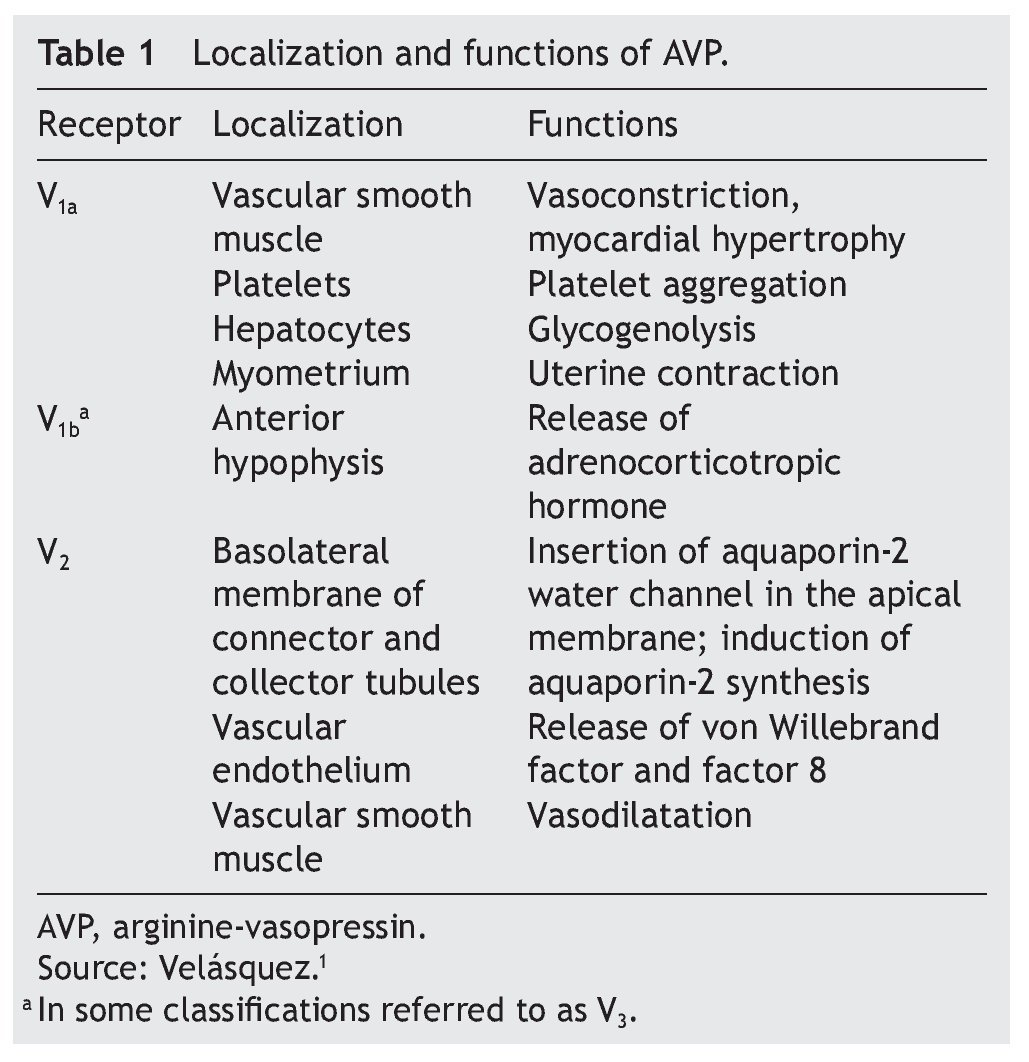
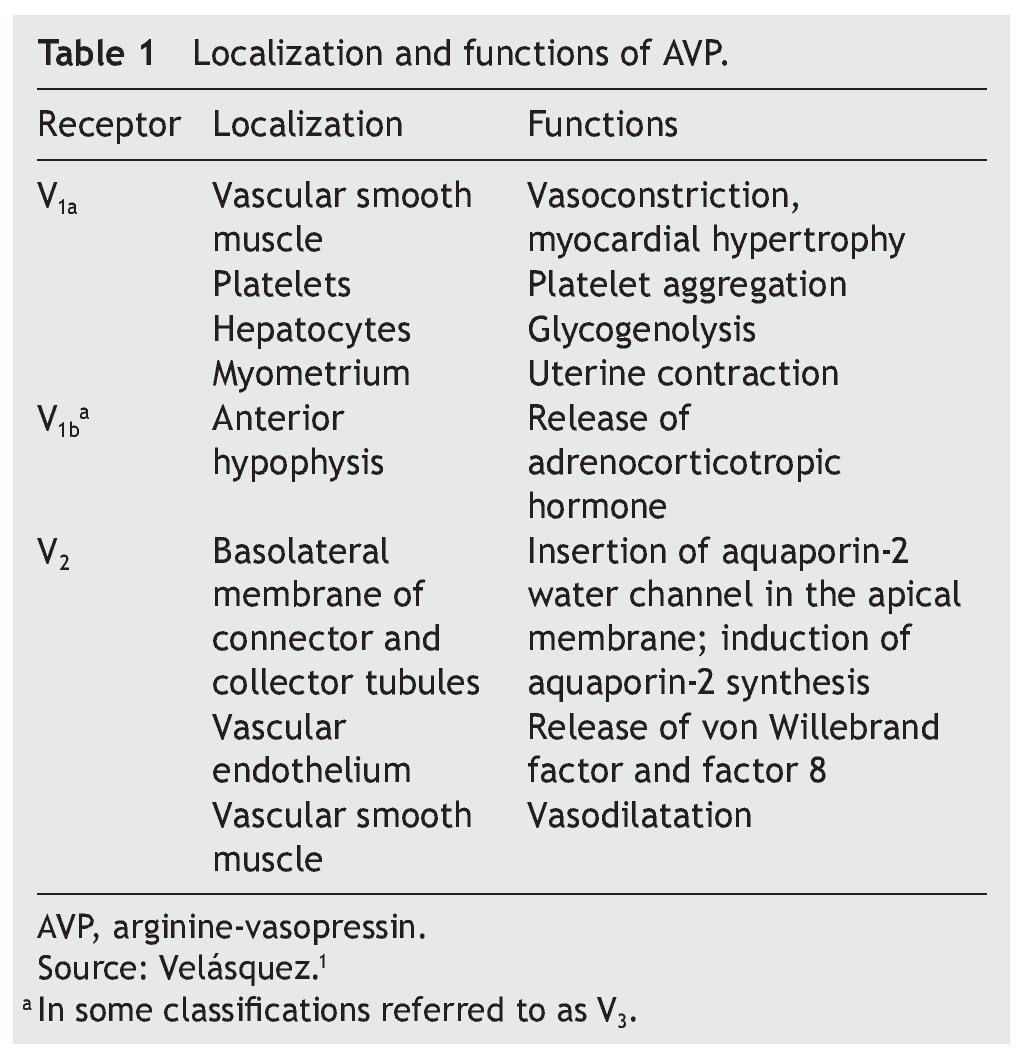
AVP receptors differ both in their location as well as their functions (Table 1).1 The V1a receptor of the AVP activates phospholipase C and increases free cytosolic calcium. The V2 receptor, which is found in the vascular endothelium and in the principal cells of the connecting and collecting tubules of the nephron, induces the release of factor 8 and von Willebrand factor and mediates the hydro-osmotic effect of the AVP. The union of AVP to the V2 receptor activates the Gs adenylcyclase system and increases the intracellular levels of cyclic 3’,5’-adenosine monophosphate (cAMP). The latter in turns activates protein kinase A, which phosphorylates the preformed aquaporin-2 water channel located in the intracellular vesicles. This phosphorylation promotes movement of the vesicles towards the apical membrane of the tubular lumen, which leads to the exocytic formation of aquaporin-2 vesicles located in the cellular membrane.2,3
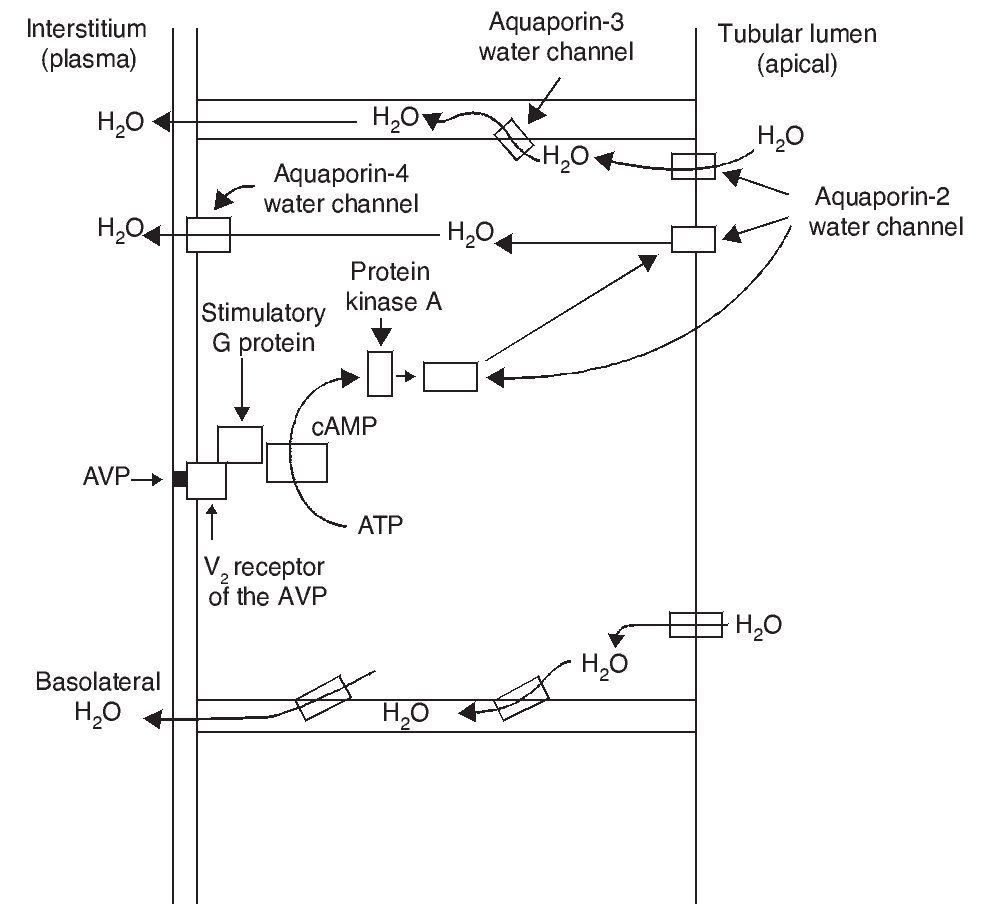
As a result of the process described, the renal tubular cell membrane that faces the tubular lumen, which is normally impermeable to water, becomes permeable. Thus, under the influence of the osmotic gradient of sodium, water is reabsorbed intracellularly, enters the cell through the aquaporin-2 water channel and exits the cell towards the interstitium through aquaporin-3 and aquaporin-4, which is located in the basolateral cell membrane (Fig. 1).1 Once the effect of the AVP ends, endocytosis of the water channels takes place, restoring water impermeability of the apical or luminal membrane.4
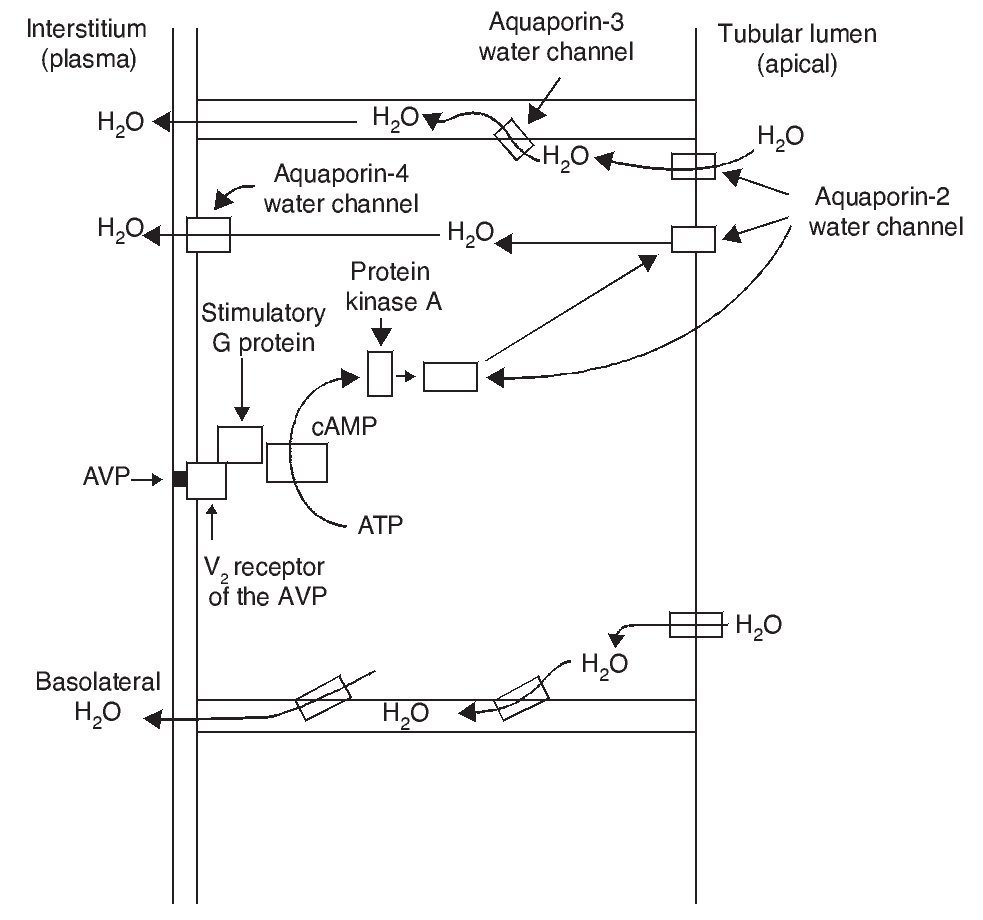
Figure 1 Action pathways of arginine vasopressin (AVP) in the collector tubule of the nephron cells. cAMP, cyclic AMP.
Aquaporins are a family of protein water channels. The first aquaporin was identified in the erythrocytes in 1991 and was called aquaporin-1.5 In the last 20 years at least 13 aquaporins have been described in mammals.6 Eight aqua-porins are expressed in the kidney, five of them–aquaporins 1, 2, 3, 4 and 7–play an important role in the regulation of body fluid balance. In particular, aquaporin 2 is regulated by AVP.7 AVP has a variety of less important actions among which are arteriolar vasoconstriction, myocardial hypertrophy, induction of platelet aggregation, glycogenolysis and release of adrenocorticotropic hormone (Table 1).3
2. Causes
As previously mentioned, nephrogenic diabetes insipidus is a clinical syndrome due to a defect or resistance of the renal tubules to concentrate urine at the stimuli of normal plasma concentrations or even elevation of AVP.8 It is classified as primary or secondary. The primary or congenital form is hereditary. The secondary form is what is observed as part of the clinical picture of different nephropathies (Table 2).

2.1. Congenital nephrogenic diabetes insipidus
In ~90% of patients with congenital nephrogenic diabetes insipidus the mode of inheritance is X-linked recessive.9 These patients have inactive mutations in the gene that codifies the V2 receptor of the AVP (AVPR2); this gene has been localized in the Xq28 region. These mutations lead to intracellular entrapment of the receptor and to the impossibility of reaching the cellular membranes in contact with the plasma. Currently, >220 different mutations of the gene that codifies the AVPR2 have been described.10-13 Almost all patients in this variant are males. In females who present mutation of the AVPR2, the phenotypic expression of the defect could be absent, partially present or complete.12
Approximately 10% of patients have an autosomal recessive inheritance. In these cases, mutations of the gene that codify the action of aquaporin-2 (AQP2) have been observed, which conditions the lack of response of the principal cells of the collecting tubules of the nephron to the action of the AVP.12,14
Finally, members of some families with the dominant autosomal variant of diabetes insipidus have been studied, in whom mutations of the gene of the aquaporin-2 has also been identified.15 It is present in <1% of patients with congenital nephrogenic diabetes insipidus. These patients characteristically present with a less severe clinical form of diabetes insipidus.12
2.2. Secondary nephrogenic diabetes insipidus
Anatomic changes in the renal medulla are frequently seen in patients with primary tubulointerstitial nephropathies, which modify the osmolar gradient dependent on the action of the mechanisms of the multiplication of the countercur-rent and cause the development of polyuria.1
In this manner, clinical scenarios of nephrogenic diabetes insipidus have been described in patients with juvenile nephronophtisis (before the development of chronic renal failure), in patients with polycystic kidney disease, distal renal tubular acidosis, Fanconi syndrome, idiopathic hypercalciuria and renal amyloidosis.1,16-18
Occasionally, some uropathic patients have hypostenouric polyuria not dependent on solute loads for prolonged periods, after the surgical correction of the urinary obstruction. Frequently renal dysplasia and chronic renal failure is seen in these patients.1 In patients with urinary tract obstruction the decrease of the expression of the aquaporins 1 to 4 has been demonstrated, and of the function of the principal sodium transporters at the renal tubular level (Na-K-ATPase, NKCC1, NCC).12
Among the different groups of patients with nephrogenic diabetes insipidus due to a secondary interstitial tubular disease, one of the most important is that caused by kaliopenic nephropathy which presents in children with severe malnutrition. It has been able to be demonstrated that in more than 75% of malnourished children with kaliopenia there was an inadequate oliguric response during states of acute dehydration.1 Renal histological study shows lesions characterized by vacuolar degeneration of the epithelial cells of the proximal convoluted tubule. There is a reduction of the sodium concentration and total solutes in the interstitium of the renal medulla in these patients. Reduction of the expression of aquaporin-2 and of the function of the sodium transporters in the renal tubule: NKCC2, NCC, ENaC (epithelial sodium channels) has also been demonstrated. A similar alteration in the functioning of the renal tubule has been seen in patients with hypercalcemia who develop the picture of nephrogenic diabetes insipidus.12 Clinical pictures of hypercalcemia associated with nephrogenic diabetes insipidus have been described in pediatric patients with vitamin D poisoning.19 The defect of the capacity of urine concentration usually persists until the baseline metabolic problem is corrected.
During a crisis episode in patients with anemia of falciform cells an increase in the viscosity of the blood may occur in the vasa recta of the renal medulla, altering the multiplier or interchange mechanisms of the counter current, decreasing the hypertonicity of the renal medulla and causing polyuria with resistance to the antidiuretic hormone.1 Similarly, the development of nephrogenic diabetes insipidus has been observed in up to 12% of patients with HIV infection who develop tubulointerstitial nephropathy.20
Diverse medications affect the renal capacity for concentrating urine and cause variable degrees of polyuria. The alteration induced by these substances is caused by inhibition of the adenylcyclase and cAMP activity in the collecting tubule, and is usually reversible when the medication is stopped.
However, alterations in the regulation of aquaporin-2 have been observed in patients who receive prolonged treatment with lithium salts (for example, treatment of bipolar disorder), the epithelial sodium channel and the urea transporters (UT-A1 and UT-B), with lesion and loss of the main cells of the collecting tubule of the nephron, which can produce an irreversible lesion of the mechanism of urine concentration.21,22 On the other hand, in patients who require short treatment with lithium salts, complete recovery of the tubulorenal alteration can be seen.23
3. Clinical manifestations
Newborns with congenital diabetes insipidus usually have normal weight at birth, although some pregnancies are occasionally complicated with polyhydramnios. The defect of urine concentration is present from birth so that the clinical manifestations could be observed from the first weeks of life. Newborns fed with mother’s breast milk may not present with early episodes of dehydration because human milk has a low salt and protein content, and therefore, low osmolar load. With the initiation of formula administration with cow’s milk, the osmolar load to the kidney increases and increases the demand for free water, which may not be supplied by the foods consumed. Therefore, the episodes of hypernatremic dehydration begin.12
Characteristically, polyuria and polydipsia are present as predominant symptoms which, as mentioned, can begin in very early ages, even from the newborn stage in the hereditary forms.
Polyuria is defined as the passage of urine in volumes >3 ml/kg/h or 90-100 ml/m2/h. The latter represents urine volumes >2,500 ml/m2/24 h or 50 ml/kg/24 h.1
In early presentations, the infant presents with persistent crying and irritability, stopping with ingestion of water or diluted milk. Also observed are vomiting, constipation and lack of weight or height gain due to decreased ingestion of nutrients as a result of the polydipsia.9 During the evolution of the disease, repeated episodes of severe dehydration may present, commonly of the hypernatremic type, accompanied by periods of weight loss, fever, seizures, and even coma. Other manifestations include constipation, nicturia and noctural enuresis in older children. Mental delay, when it presents itself, is a consequence of the repeated episodes of hypernatremic dehydration and the very energetic rehydration treatments that can cause cerebral edema.24 The presence of intracranial calcifications, more frequent in children who already have mental retardation, probably a result of episodes of bleeding and necrosis, have also been described.12
In older children, intense thirst is associated with polyuria with volumes of various liters, and that on occasion is manifested by enuresis, growth delays and anorexia occurs, with preference for intake of cold water. The foregoing precludes progressive weight loss. Patients present apathy and easy irritability, and their performance in school is poor.1
Persistent polyuria can cause the development of mega bladder, hydroureter and hydronephrosis.1,24 It has been observed in some children that high urinary flow causes the development of a trabeculated bladder in the absence of an infravesical obstruction. Urodynamic studies show a complacent bladder with slow and incomplete emptying, which exacerbates deterioration of renal function for which procedures (such as cystostomy) have been required to ensure bladder emptying.25 On the other hand, there have been reports of patients with nephrogenic diabetes insipidus who also present lower urinary tract obstruction (inferior urethral valves). These cases allow to emphasize that studies of patients with nephrogenic diabetes insipidus and hydronephrosis should also rule out aggregated urinary obstructive causes.26,27
4. Diagnosis
Generally, urinary osmolality ranges from 50-200 mOsm/ kg H2O and urine density between 1.001 and 1.005. It has been mentioned that urinary volumes are usually >2,500 ml/ m2/24 h clear and colorless urine.
Increase of serum concentrations of sodium, chloride and urea can be seen in the blood due to the negative balance of water and a tendency to serum hyperosmolality.
Under these conditions, it is advisable to perform a test of urinary concentration, which consists of stimulating the maximum renal concentration in response to water restriction. This test is very useful to establish the diagnosis of diabetes insipidus of any etiology and to differentiate from compulsive polydipsia or potomania. In the former, the patient generally, because of psychological disorders, ingests large quantities of water and other fluids, which causes a compensatory polyuria and a clinical picture similar to diabetes insipidus is seen, but without completely affecting the power of renal concentration. This picture has been described even in infants.1
The fluid deprivation test should be performed in young children with significant polyuria and possible diagnosis of primary neurogenic or nephrogenic diabetes insipidus, under strict vigilance so as to avoid a severe picture of dehydration.
The procedure for the test is described below.1,16
a. The test is begun after 8 a.m. with strict weight monitoring after each episode of urination and of clinical signs of dehydration. Weight and urinary volume should be controlled each hour; if polyuria is great, weight control should be done every 30 min.16 Osmolality and density should be determined for the urine samples obtained. When the patient has lost 3% of his initial weight, a sample should be collected for serum and urinary osmolality as well as for urine density.
b. Under normal conditions the increase in urinary osmolality with levels >800 mOsm/kg H2O or densities of 1.020 or greater in children and adolescents should be observed. In infants osmolality >500 mOsm/kg H2O or density >1.015 should be observed.28
c. When the concentration test has revealed a deficiency of the ability of the renal concentration, the AVP response test should be performed. With this test, neurogenic diabetes insipidus will be differentiated from nephrogenic. The AVP test consists of the subcutaneous administration of aqueous vasopressin11 at a dose of 10 mU/kg or 1 U/ m2 or of 1-desamino-8-D-arginine vasopressin (DDAVP), the nasal biosynthetic antidiuretic hormone, at a dose of 10 mg for pre-school age children and 20 mg in school-age children followed by a 2-h urine collection. With these urine samples the osmolality and density and the amount of urine will be determined.
d. The volume reduction and increase in osmolality and urine density after the administration of vasopressin are indicative of the renal concentration capacity and allow for the diagnosis of central diabetes insipidus or neurogenic diabetes insipidus to be made (the normal response takes the urinary osmolality to >800 mOsm/kg H2O or >50% of the baseline osmolarity16 with a density >1.020). As an example, if the dehydration test led to the production of urine with an osmolality of 200 mOsm/ kg H2O and with the application of AVP it increased to >800 mOsm/kg H2O or >50% from the baseline osmolarity, the diagnosis of neurogenic or central diabetes insipidus is established.
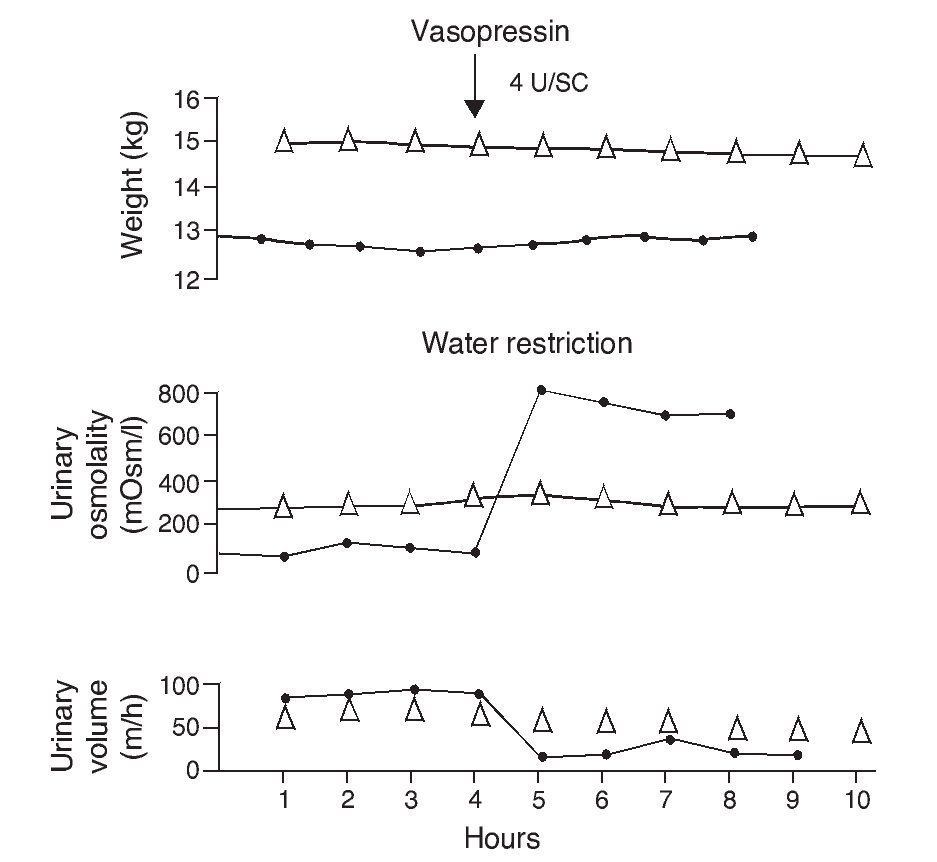
e. In contrast, the persistence of a lack of response corresponds to cases of nephrogenic diabetes insipidus (Fig. 2).1,29 A slight increase of the urinary osmolality can occur in these patients, but this increase is usually <150 mOsm/kg H2O or <20% of the baseline values.1,16 Uri-nary osmolality usually remains <200 mOsm/kg H2O.In patients with secondary nephrogenic diabetes insipidus, the urinary osmolarity obtained after the administration of AVP usually is greater than what is seen in children with congenital nephrogenic diabetes insipidus.12
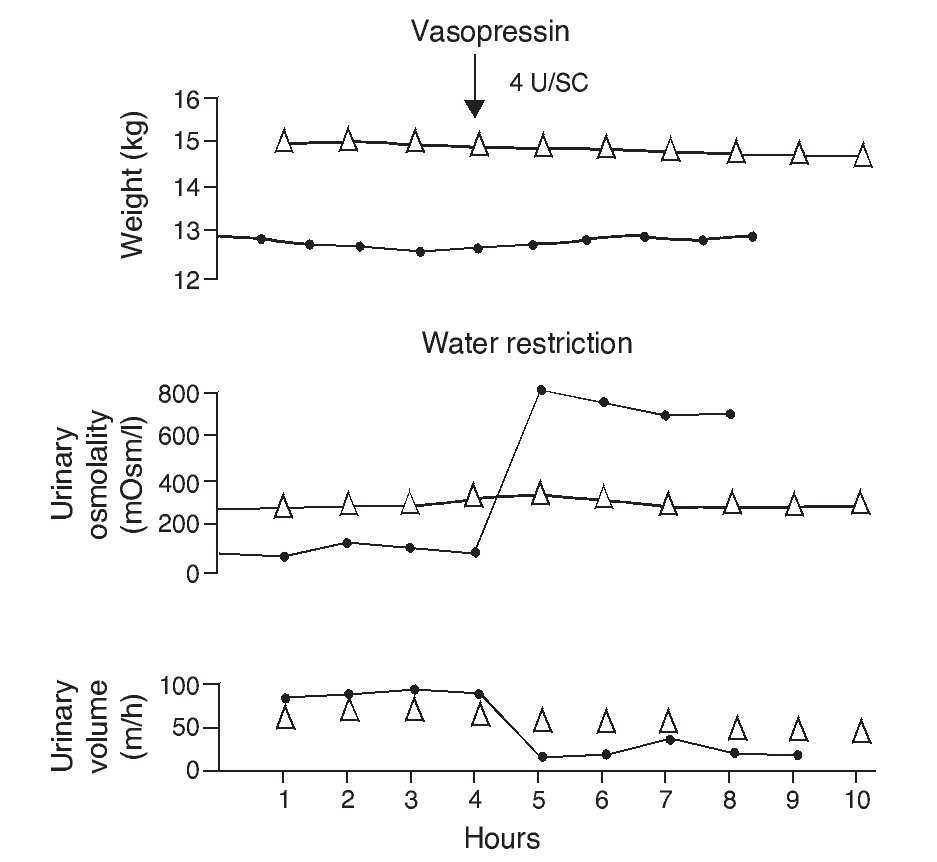
Figure 2 Urine concentration test in two children with diabetes insipidus. The patient with neurogenic diabetes insipidus (continuous line with black circles) presented volume decrease with increase in urine osmolality after administration of vasopressin. On the other hand, there was no response in the patient with nephrogenic diabetes insipidus (line with triangles). Source: Dorantes.29 Reproduced with permission.
4.1. Plasma levels of AVP
A direct correlation between the plasma levels of AVP determined by radioimmunoassay and plasma osmolality after a concentration test in normal subjects has been demonstrated. Inadequately low levels for plasma osmolality have also been demonstrated in patients with neurogenic diabetes insipidus and high levels of vasopressin in those with nephrogenic diabetes insipidus.1,29
5. Treatment
There is no specific treatment when we are dealing with a primary disorder. Therefore, fluid intake is fundamental to prevent the deleterious effects of repeated episodes of dehydration. Due to risk of developing extended urinary bladders due to polyuria, the bladder should be emptied regularly to ensure the maximal bladder capacity is maintained within the normal range.
Infants frequently cannot drink sufficient fluids to compensate for urinary losses. In these cases, it is useful to reduce the sodium content in the diet (1 mEq/kg/day) so as to decrease the solute load. It is not advisable to reduce the protein content because it can lead to malnutrition.1
Diuretics, such as hydrochlorothiazide (1 to 2 and up to 4 mg/kg/day), were the first class of medications effective in reducing the urinary volume. When combined with the reduction in salt intake, hydrochlorothiazide can reduce uri-nary volume from 20 to 50% of baseline values. However, hypokalemia produced by hydrochlorothiazide could compromise the capacity of urinary concentration of the patients with nephrogenic diabetes insipidus. Therefore, it is necessary to administer a potassium supplement.1,29
Currently there is sufficient evidence to continue treatment with hydrochlorothiazide and amiloride (0.3 mg/ kg/day) plus a potassium-sparing diuretic or an inhibitor of prostaglandin synthesis such as indomethacin (2 mg/ kg/24 h). Because of the elevated frequency with which indomethacin causes side effects such as gastrointestinal complications (anorexia, nausea, vomiting, abdominal pain, ulceration and intestinal perforations, hemorrhage), hematological changes (neutropenia, thrombocytopenia, anemia) and renal dysfunction, the first combination has been suggested as the treatment of choice in these patients, particularly in children 4 to 6 years of age. Younger children do not tolerate amiloride well due to the persistent feeling of nausea, for which it is probable they would require the combination with indomethacin in the first years of life.1,12,29
It has been suggested that the apparent paradoxical effect of hydrochlorothiazide occurs due to, in the absence of vasopressin arginine, the luminal hydrochlorothiazide increases the osmotic and dilutional permeability of water in the collecting tubules of the nephron that cross the internal renal medulla through independent stimulation of AVP of aquaporin-2.10,30 It has also been proposed that the hypovolemic state induced by the diuretic effect of the thiazides by inhibition of the sodium-chloride co-transporter in the renal tubules stimulates the renin-angiotensin II-aldosterone system, which causes greater reabsorption of sodium and water (via aquaporin-1) in the proximal tubules of the nephron. When prostaglandins are administered, the effect of the thiazides decreases. This may explain why indomethacin enhances the effect of the thiazides in patients with nephrogenic diabetes insipidus.1,30
Studies for the treatment of congenital nephrogenic diabetes insipidus have been recently done through drug stimulation, promoting the transport of the AVPR2 retained in the cytoplasm of the tubulorenal cell to the plasma membrane where it could develop its function. Other studies are directed at achieving the direct stimulation of the retained AVPR2 or directly stimulate the function of aquaporin-2 without the need of the participation of AVPR2.12,21,31-36
On the other hand, different studies have been published in which the effect of the prostaglandin E2 or the related agonists has been investigated (such as the EP2 and EP4 prostanoid receptors) in experimental animals. These induce, paradoxically, increase in the activity of cAMP, independently of the action of the AVP, with the phosphorylation of aquaporin-2 and its insertion in the tubular membrane of the nephron.2,34 This could provide a new treatment modality in patients with chromosome X-linked nephrogenic diabetes insipidus.
Conflict of interest
The authors declare no conflict of interest of any nature.
Received 14 July 2014;
accepted 14 October 2014
☆ Please cite this article as: Velásquez-Jones L, Medeiros-Domingo M. Diabetes insípida nefrogénica. Bol Med Hosp Infant Mex. 2014;71:332-338
* Corresponding author.
E-mail: velasquezjones@hotmail.com (L. Velásquez-Jones).