


El raquitismo dependiente de vitamina D tipo I es una enfermedad hereditaria rara debida a una mutación en el gen CYP27B1 que codifica la enzima 1α-hidroxilasa. Se caracteriza por la presentación de raquitismo hipocalcémico grave desde la edad de la lactancia debido al déficit de producción del metabolito activo de la vitamina D, la 1α,25-dihidroxivitamina D3.
Caso clínicoPresentamos el caso de un paciente con raquitismo diagnosticado a los 11 meses de edad y el seguimiento hasta los 9 años.
ConclusionesSe discute la fisiopatología de la enfermedad y la importancia del diagnóstico y tratamiento oportunos.
Vitamin D dependent rickets type I is a rare hereditary disease due to a mutation in CYP27B1 encoding the 1α-hydroxylase gene. Clinically, the condition is characterized by hypocalcemic rickets in early infancy due to a deficit in the production of the vitamin D active metabolite 1,25-dihydroxy-vitamin D3.
Case reportWe report the case of a patient diagnosed at 11 months with follow-up until 9 years of age.
ConclusionsThe pathophysiology of the disease and the relevance of early diagnosis and management are discussed.
En 1961, Prader y colaboradores describieron una forma de raquitismo hereditario a la cual denominaron “raquitismo por seudodeficiencia”1. Este tipo de raquitismo podía diferenciarse de la hipofosfatemia ligada al cromosoma X debido al modo diferente de transmisión, a la presencia de hipocalcemia grave y a la posibilidad de remisión con dosis altas de vitamina D. En 1973, Fraser y colaboradores trataron a un paciente con este tipo de raquitismo con dosis fisiológicas de la forma activa de la vitamina D, la 1α-25-dihidroxivitamina D3 [1α,25(OH)2D3], por lo que sugirieron que esta enfermedad era consecuencia de un defecto de la enzima 25(OH)D3-1α-hidroxilasa en el riñón2. Posteriormente, en 1978, Brooks y colaboradores describieron un paciente con características clínicas similares, pero con niveles elevados de la 1α,25(OH)2D3 en suero antes y durante el tratamiento. Por lo referido previamente, estos autores sugirieron que el raquitismo dependiente de vitamina D podía ser clasificado como tipo I [deficiente producción de la 1α,25(OH)2D3] o tipo II [falta de respuesta del órgano blanco a la 1α,25(OH)2D3].3
El raquitismo dependiente de vitamina D tipo I es una enfermedad muy rara. En la experiencia del Departamento de Nefrología del Hospital Infantil de México Federico Gómez se han estudiado cuatro pacientes en los últimos 38 años.4,5 Por lo anterior, consideramos de interés presentar las características clínicas, la respuesta al tratamiento y la evolución a largo plazo de un niño en quien se realizó el diagnóstico de raquitismo dependiente de vitamina D tipo I a la edad de 11 meses y se continuó su tratamiento y control hasta los 9 años de edad.
2Caso clínicoLactante de 11 meses de edad, sexo masculino, padre de 30 años y madre de 33 años, aparentemente sanos. Un hermano de 10 años sano y un hermano fallecido al año de edad por infección neumónica con diagnóstico de raquitismo.
Producto de parto eutócico, peso al nacer 3.6kg, recibió seno materno hasta los 7 meses de edad, ablactación a los 6 meses. Sostén cefálico a los 3 meses, sedestación a los 9 meses de edad.
Ingresó por cuadro clínico caracterizado por tos de 2 semanas de evolución, dificultad respiratoria de 7 días y fiebre de 3 días de evolución. Al ingreso su peso fue de 7.6kg (valor Z −3.33) y la talla de 65cm (valor Z −3.72), la presión arterial 90/60mm Hg. Presentó hipotonía muscular generalizada, cráneo de conformación normal, tórax en quilla, rosario costal bilateral, campos pulmonares con estertores crepitantes bilaterales, retracción xifoidea, hígado palpable a 3cm por debajo del borde costal, ensanchamiento de articulaciones de codos, muñecas, rodillas y tobillos.
La radiografía de tórax mostró infiltrado parahiliar bilateral por lo cual, con el diagnóstico de neumonía adquirida en la comunidad, se indicó tratamiento antibiótico. Presentó evolución favorable.
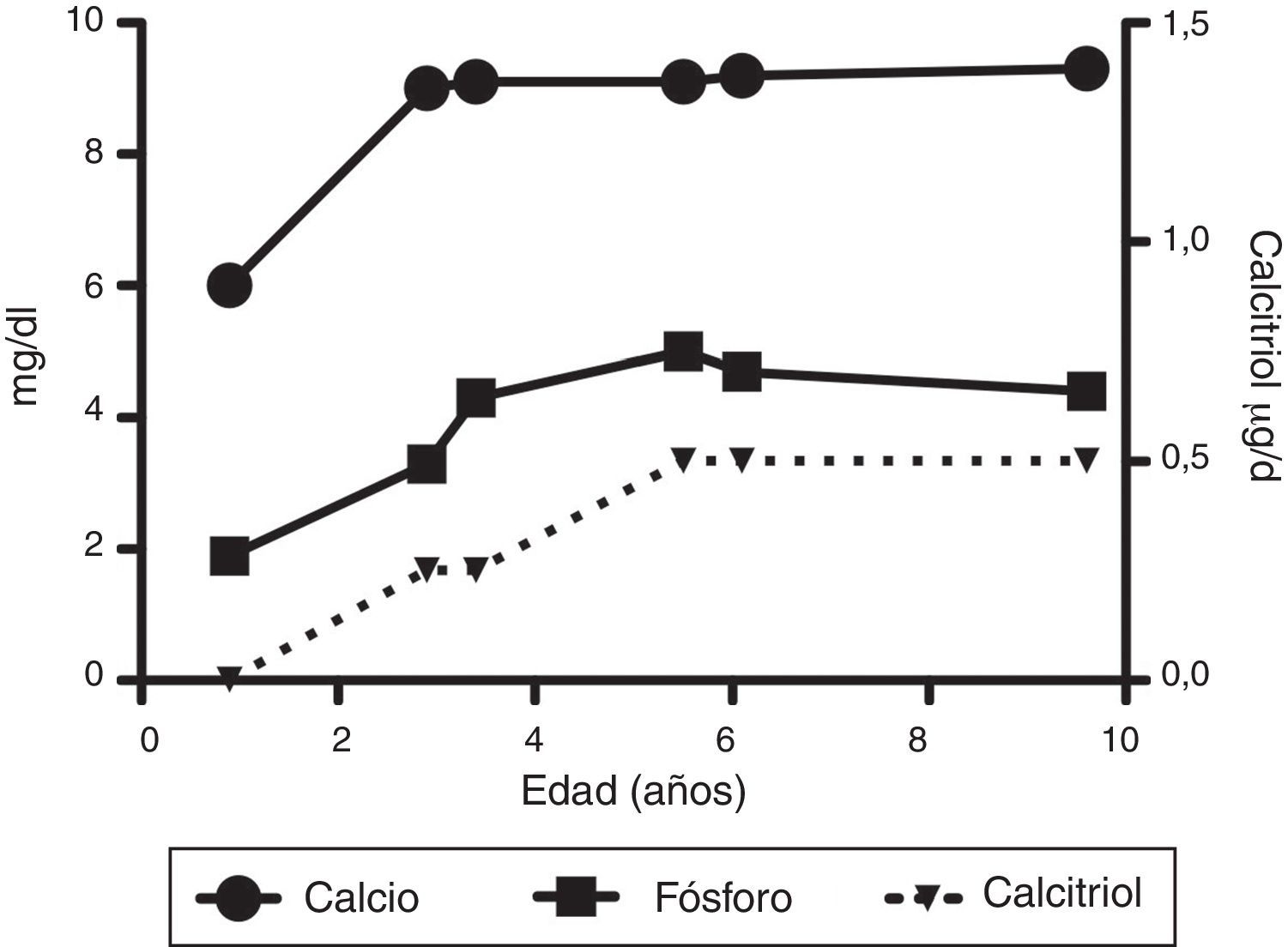
Los exámenes en sangre realizados durante su internamiento mostraron los siguientes resultados: hemoglobina 12g/dl, glucosa 98mg/dl, sodio 139mmol/l, potasio 4.1mmol/l, CO2 total 22 mEq/l; proteínas totales 6.6g/dl, albúmina 3.6g/dl, calcio 6.0mg/dl, fosfato 1.9mg/dl, magnesio 1.8mg/dl, fosfatasa alcalina 466 U/l, creatinina 0.1mg/dl; bilirrubinas y aminotransferasas normales; hormona paratiroidea 571pg/dl (normal 9-25pg/dl); 1α,25-(OH)2D3 < 5pg/ml (normal 15 a 90pg/ml); 25-hidroxicolecalciferol [25(OH)D3] 23.3ng/ml (normal 17 a 54ng/ml).
Los exámenes de orina y función renal mostraron calciuria de 0.71mg/kg/d (normal<4mg/kg/d); relación calcio/creatinina 0.1mg/mg (normal para la edad<0.6mg/mg); fosfaturia 25.6mg/kg/d (normal<20mg/kg/d); reabsorción tubular de fosfatos del 87% (normal>80%). Examen general de orina: pH 7.0, densidad 1.024, albúmina negativa, glucosa negativa, resto normal. El ultrasonido renal mostró dimensiones normales de la silueta renal para su edad sin datos de nefrocalcinosis.
El estudio radiográfico de los huesos largos tanto en miembros superiores como en miembros inferiores mostró desmineralización ósea, ensanchamiento de las epífisis e irregularidades de las metáfisis con deshilachamiento y, en algunos de ellos, evidencia de “copa invertida”. En miembros superiores, además, se observó fractura patológica del húmero del brazo derecho. Los estudios de calcio, fosfato y fosfatasa alcalina realizados en los padres fueron normales.
Diez días después de su ingreso ya con el diagnóstico de raquitismo dependiente de vitamina D tipo I, se inició manejo con calcitriol (1α,25(OH)2D3) 0.25μg cada 24h, vía oral y solución de fosfatos 2ml (60mg) cada 6h (35mg/kg/d). Una semana después, por persistencia de hipocalcemia grave (5.9mg/dl), se incrementó el calcitriol a 0.5μg cada 24h, solución de fosfatos 4ml cada 6h y se inició carbonato de calcio 195mg cada 6h. Se administraron varias infusiones de gluconato de calcio por vía intravenosa por hipocalcemia persistente.
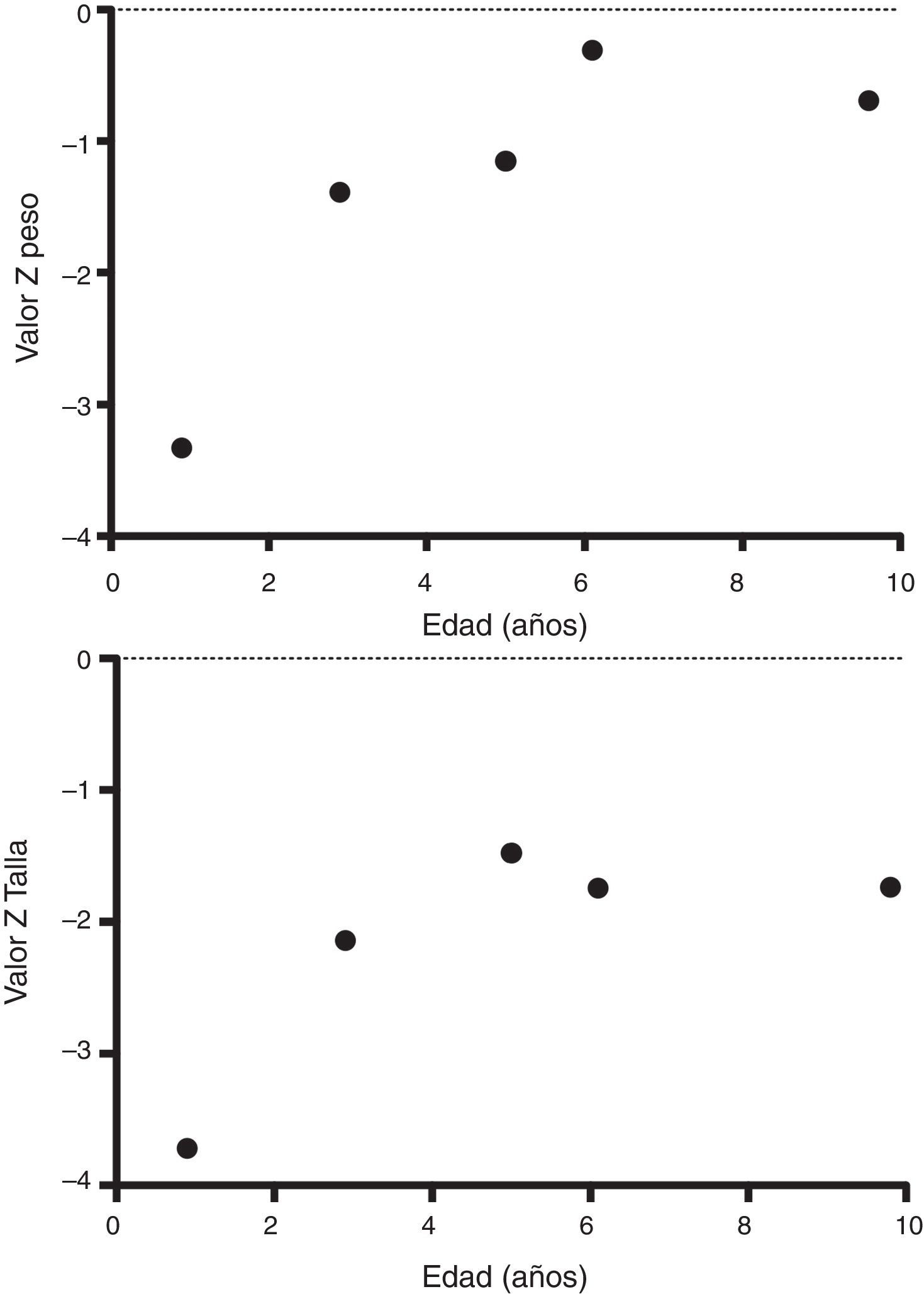
A su egreso, 40 días después, los exámenes en sangre mostraron sodio de 139mmol/l, potasio 4.5mmol/l, calcio 7.1mg/dl, fósforo 3.3mg/dl y magnesio 2.2mg/dl. En ese momento se encontraba recibiendo calcitriol 0.75μg cada 12h, solución de fosfatos 6ml cada 6h y carbonato de calcio 1g cada 6h. Hubo una mejoría notable en su crecimiento durante el primer año del inicio del tratamiento (Figura 1), que se ha mantenido a lo largo del seguimiento ajustando el aporte de calcitriol con el fin de llevar las cifras de calcio y fosfato séricos a valores normales (Figura 2).


En su control posterior se registró el inicio de la deambulación a los 15 meses de edad, y a los 2 años 9 meses de edad presentó peso de 12.8kg (valor Z −1.39) y talla 80cm (valor Z −2.14). Se encontraba recibiendo tratamiento solamente con calcitriol a dosis de 0.25μg/d. Los resultados de laboratorio mostraron calcio 9.8mg/dl, fosfato 2.9mg/dl creatinina 0.2mg/dl; el examen general de orina: pH 6.5, densidad urinaria 1.028, resto negativo. Se realizó un estudio radiográfico de control de la muñeca derecha que mostró mejoría importante de las lesiones óseas, con edad ósea de 1 año 9 meses.
A los 5 años de edad presentó peso de 16.9kg (valor Z −0.9) y talla de 102cm (valor Z −1.16). Debido al descenso progresivo de la concentración de calcio en el suero (8.0mg/dl) se incrementó la dosis de calcitriol a 0.5μg/d. El calcio sérico se normalizó a 9.1mg/dl después de 6 meses.
A la edad de 9 años se observó peso de 25.1kg (valor Z −1.36) y talla de 126cm (valor Z −1.74). Los exámenes de laboratorio en suero mostraron creatinina 0.4mg/dl, calcio 9.4mg/dl, fósforo 4.2mg/dl, fosfatasa alcalina 175 U/l y hormona paratiroidea 25.1pg/ml. El estudio radiográfico de huesos largos no mostró signos de raquitismo. Se indicó mantener el tratamiento con calcitriol en dosis de 0.5μg/d.
3DiscusiónEl raquitismo dependiente de vitamina D tipo I (OMIM 264700)6,7 también se ha denominado “dependencia hereditaria de vitamina D tipo I”, “seudodeficiencia hereditaria de vitamina D” y “deficiencia hereditaria selectiva de 1α,25(OH)2D3”.8
Este tipo de raquitismo se transmite en forma autosómica recesiva. El gen responsable ha sido identificado como CYP27B1. Se encuentra localizado en el cromosoma 12q13.1-13.3 y codifica a la enzima 25(OH)D3-1α-hidroxilasa.6,9,10 La disponibilidad de la información de la secuencia del gen CYP27B1ha permitido investigar las mutaciones en los pacientes con esta enfermedad y en los portadores obligados11,12. Se han identificado más de 60 mutaciones en diferentes grupos familiares estudiados12–16. La presencia de estas mutaciones provoca la supresión de la actividad de la 25(OH)D3-1α-hidroxilasa, y por consiguiente el déficit en la síntesis de la forma activa de la vitamina D, la 1α,25(OH)2D3.9,12
Los pacientes afectados con esta forma de raquitismo presentan manifestaciones clínicas tempranas relacionadas con la hipocalcemia grave.5 Así, este paciente con apariencia normal al nacimiento a los 11 meses de edad presentaba alteraciones óseas que incluyeron fracturas patológicas en el brazo derecho, deformidad de la parrilla costal (“tórax en quilla”), rosario costal y ensanchamiento de las epífisis óseas, sobre todo en la articulación de la muñeca, pero también aparente en codos, rodillas y tobillos. La deformación de la caja torácica en estos pacientes puede condicionar una reducción de la capacidad respiratoria y poner en peligro la vida en caso del desarrollo de una complicación infecciosa del parénquima pulmonar. De hecho, en la historia clínica se refirió que un hermano del paciente, también con manifestaciones clínicas de raquitismo, falleció al año de edad por neumonía. Por su parte, el paciente estudiado ingresó al hospital a consecuencia de un cuadro infeccioso neumónico.
Otros pacientes estudiados han presentado tempranamente (antes del año de edad) crisis convulsivas a consecuencia de la hipocalcemia grave. En estos casos y en presencia de manifestaciones raquíticas debe sospecharse la posibilidad de raquitismo dependiente de vitamina D.5,16,17
Al igual que se observó en el paciente estudiado, se ha descrito la presencia de hipotonía muscular, lo cual condiciona retraso en el desarrollo de actividades motoras en estos niños.7,17
En niños con raquitismo dependiente de vitamina D, además de la presencia de hipocalcemia grave, puede encontrarse elevación de la concentración sérica de la hormona paratiroidea con desarrollo de hiperparatiroidismo secundario.6 Habitualmente, como consecuencia de lo anterior y como se observó en este paciente, los niveles de fosfato en el suero se encuentran disminuidos. Además, en algunos pacientes puede observarse también pérdida tubular proximal de bicarbonato (con desarrollo de acidosis metabólica hiperclorémica) y aminoaciduria generalizada.18 Las alteraciones anteriores obligan a realizar el diagnóstico diferencial con dos desórdenes tubulares renales: la hipofosfatemia ligada al cromosoma X y el síndrome de Fanconi, sobre todo en su variedad de cistinosis nefropática del lactante.19 Estas alteraciones tubulares renales se normalizan habitualmente en los primeros 2 meses de haber iniciado el tratamiento con calcitriol en relación con la normalización de los niveles en suero de la hormona paratiroidea.18
Se ha mencionado que en los pacientes con raquitismo dependiente de vitamina D tipo I se han demostrado defectos en la producción de la 1α,25(OH)2D3.9 Por consiguiente, el diagnóstico de raquitismo dependiente de vitamina D tipo I se basa en el hallazgo de niveles muy reducidos o no detectables de la 1α,25(OH)2D3.16 Esto permite realizar el diagnóstico diferencial con el tipo II, en el cual se encuentran niveles elevados de este metabolito.20 Este último tipo de raquitismo se ha denominado raquitismo hereditario resistente a la vitamina D.20 Por otro lado, los niveles de 25(OH)D3 pueden encontrarse normales o ligeramente aumentados,16 como ocurrió en el paciente motivo de este informe.
En el estudio radiográfico de los huesos largos se observa ensanchamiento y distorsión de la placa de crecimiento con irregularidad en la zona de mineralización de las metáfisis con apariencia de deshilachamiento (forma de “copa invertida”). La corteza ósea se encuentra muy delgada con escasas zonas trabeculares. Son frecuentes las seudofracturas. En algunos niños gravemente afectados puede observarse deformación torácica (“tórax en quilla”).
Los pacientes con raquitismo dependiente de vitamina D tipo I han sido tratados con análogos del calciferol, como el α-calcidiol o 1α-hidroxicolecalciferol [1α-(OH)D3], el cual es convertido a 1α,25(OH)2D3 por acción de la 25-hidroxilasa en el hígado.4,6,16,17 Otro tratamiento utilizado es la administración del metabolito activo de la vitamina D deficiente, la 1α,25(OH)2D3.5,7,18,20 Se requieren habitualmente dosis iniciales altas de la 1α,25(OH)2D3 (entre 0.5 y 3μg/d). Estas dosis deberán ser ajustadas posteriormente para mantener los niveles de calcio sérico en el rango normal, como se realizó en el paciente estudiado. De esta manera, al mantener la actividad normal del principal metabolito activo de la vitamina D, también se activarán adecuadamente los receptores de la vitamina en sus órganos blanco. El receptor nuclear de la vitamina D, una fosfoproteína nuclear de 50kDa, pertenece a la superfamilia de receptores nucleares que incluye los receptores de las hormonas esteroideas y tiroideas.21,22 La pérdida renal de fosfato encontrada al diagnóstico se debe al hiperparatiroidismo secundario que se produce por la reducción en la absorción intestinal de calcio.23 El tratamiento con la 1α,25(OH)2D3 se indica de manera permanente.
El paciente estudiado requirió dosis altas de vitamina D al inicio para corregir la hipocalcemia y el hiperparatiroidismo secundario, además del aporte suplementario de calcio por vía oral en forma de carbonato de calcio.18 Al momento actual se han mantenido niveles normales de calcio en suero con dosis de 0.50μg/d. Además, debido al desarrollo de hipofosfatemia, el paciente requirió un suplemento de solución de fosfatos en las etapas iniciales del tratamiento.
En conclusión, el hallazgo de un niño lactante con manifestaciones raquíticas a consecuencia de hipocalcemia grave, en quien se ha descartado clínicamente un raquitismo carencial, debe llamar la atención a la posibilidad diagnóstica de un raquitismo, ya sea dependiente de la falta de producción del metabolito activo de la vitamina D por el riñón o de la resistencia al efecto de la misma en los órganos blanco. En el primer caso, que es la característica principal en el raquitismo dependiente de vitamina D tipo I, el tratamiento adecuado conducirá a la corrección de las alteraciones clínicas, bioquímicas y óseas de los niños afectados, mejorando su crecimiento.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
Fuente de financiamientoNinguna.
Conflictos de interésLos autores declaran no tener ningún conflicto de intereses.