






La leucemia linfoblástica aguda (LLA) es el tipo de cáncer más frecuente en niños. Aunque se sabe que las alteraciones genéticas constituyen la base de la etiología de la LLA, se ha demostrado que no son suficientes para el desarrollo leucémico; son necesarias alteraciones adicionales, como las modificaciones epigenéticas. En la LLA se han identificado alteraciones de este tipo, como la metilación del DNA, la modificación de histonas y la regulación por RNAs no codificantes. La hipermetilación del DNA en regiones promotoras es una de las alteraciones epigenéticas más frecuentes en LLA: y conlleva al silenciamiento de genes que generalmente son supresores de tumor y, en consecuencia, contribuye a la leucemogénesis. También se han detectado alteraciones en proteínas remodeladoras de histonas, como la sobreexpresión de enzimas desacetilasas de histonas, así como alteraciones en enzimas acetil transferasas y metil transferasas. En la LLA también se altera la expresión de miRNAs, lo cual produce desregulación en la expresión de sus genes blanco. Estas modificaciones epigenéticas son eventos clave en la transformación maligna, e involucran la desregulación de oncogenes como BLK, WNT5B y WISP1 y de supresores de tumor como FHIT, CDKN2A, CDKN2B y TP73, lo que afecta diversos procesos celulares fundamentales que conllevan al desarrollo de LLA. Las alteraciones epigenéticas y genéticas contribuyen en conjunto al desarrollo y evolución de la LLA.
Acute lymphoblastic leukemia (ALL) is the most common childhood cancer. It is well-known that genetic alterations constitute the basis for the etiology of ALL. However, genetic abnormalities are not enough for the complete development of the disease, and additional alterations such as epigenetic modifications are required. Such alterations, like DNA methylation, histone modifications, and noncoding RNA regulation have been identified in ALL. DNA hypermethylation in promoter regions is one of the most frequent epigenetic modifications observed in ALL. This modification frequently leads to gene silencing in tumor suppressor genes, and in consequence, contributes to leukemogenesis. Alterations in histone remodeling proteins have also been detected in ALL, such as the overexpression of histone deacetylases enzymes, and alteration of acetyltransferases and methyltransferases. ALL also shows alteration in the expression of miRNAs, and in consequence, the modification in the expression of their target genes. All of these epigenetic modifications are key events in the malignant transformation since they lead to the deregulation of oncogenes as BLK, WNT5B and WISP1, and tumor suppressors such as FHIT, CDKN2A, CDKN2B, and TP53, which alter fundamental cellular processes and potentially lead to the development of ALL. Both genetic and epigenetic alterations contribute to the development and evolution of ALL.
Las leucemias constituyen el cáncer pediátrico más común; el tipo más frecuente es la leucemia linfoblástica aguda (LLA)1. En la LLA se han identificado diversas alteraciones genéticas específicas que conllevan al desarrollo de esta enfermedad1,2; sin embargo, se reconoce que no son suficientes para la transformación leucémica, por lo que se requieren eventos adicionales3. Actualmente se sabe que además de las alteraciones genéticas, los cambios epigenéticos también son fundamentales en el proceso leucemogénico4–6.
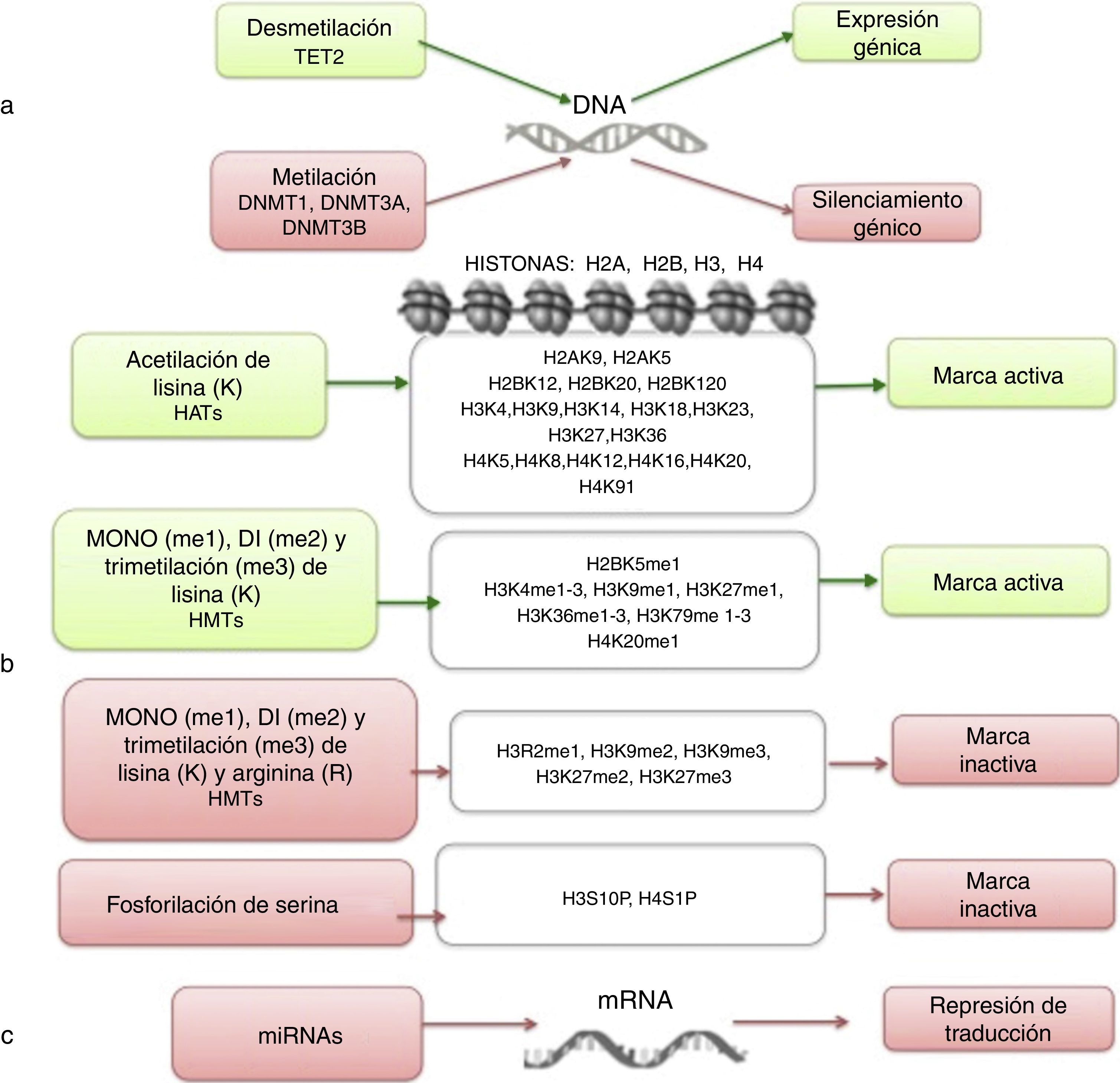
La epigenética estudia los cambios heredables que llevan a modificaciones en la expresión génica y que no implican alteraciones en la secuencia de DNA7,8. Entre los mecanismos epigenéticos más estudiados se encuentran la metilación del DNA, la modificación de histonas, la remodelación de nucleosomas y la regulación de RNAs no codificantes (fig. 1)9,10. Estas alteraciones epigenéticas son eventos fundamentales en el desarrollo de la LLA. En esta revisión se definen y presentan las principales alteraciones epigenéticas que se han detectado en la LLA. Se incluye la metilación en el DNA, las modificaciones de histonas como la acetilación, la regulación por microRNAs (miRNAs) y los genes que se modifican por estos mecanismos. Aunque las alteraciones epigenéticas son fundamentales en LLA de estirpes B y T (LLA-B y LLA-T), tanto en adultos como en niños, esta revisión se enfoca principalmente en la LLA-B pediátrica debido a la alta incidencia de esta enfermedad en este grupo etario. El estudio de las modificaciones epigenéticas de la LLA es de gran importancia para el conocimiento sobre la etiología de la enfermedad, así como para la búsqueda de blancos terapéuticos4,9.
, las modificaciones en las histonas (b) y la regulación por RNAs no codificantes (c). Se indica en cada caso bajo qué condiciones se induce expresión génica (verde) o silenciamiento (rojo).")
Niveles de regulación epigenética. La regulación epigenética involucra mecanismos como la metilación del DNA (a), las modificaciones en las histonas (b) y la regulación por RNAs no codificantes (c). Se indica en cada caso bajo qué condiciones se induce expresión génica (verde) o silenciamiento (rojo).
La LLA se caracteriza por la proliferación descontrolada de células inmaduras linfoides llamadas linfoblastos, que predominan en la médula ósea y alteran la hematopoyesis normal2,11. La LLA puede afectar la estirpe de linfocitos T o B y se presenta en adultos y niños11. En la etapa pediátrica es de especial relevancia, ya que es el tipo de cáncer más común, representando el 25% de los casos. El 85% de las LLA en niños son de subtipo precursor B (LLA-B). Las LLA pediátricas son fenotípicamente y genéticamente heterogéneas y la etiología no ha sido completamente determinada12.
La LLA ha sido clasificada en subgrupos de acuerdo con la presencia de alteraciones genéticas, las cuales tienen una fuerte influencia en el pronóstico de los pacientes13. Aproximadamente el 75% de los casos de LLA pediátrica presentan alguna alteración cromosómica numérica o estructural14. La alteración numérica más común en la LLA pediátrica es la hiperdiploidía15; las alteraciones cromosómicas estructurales más frecuentes son las translocaciones16. La translocación t(12;21)(p13;q22) genera la fusión de los genes ETV6-RUNX1 y se presenta del 20-25% de los casos15,16. La translocación t(1;19)(q23;p13) conlleva a la fusión de los genes TCF3-PBX1 y se presenta del 5-6% de los casos de LLA pediátrica13. La translocación t(9;22)(q34;q11) genera la fusión de los genes BCR-ABL1 y se presenta en el 3% de los casos11,16. Las translocaciones que involucran al gen MLL (KMT2A) se detectan en el 3% de los pacientes de entre 2-5 años de edad y en el 80% de los menores de un año. Aunque estas alteraciones genéticas son capaces de iniciar el proceso de leucemogénesis, generalmente no son suficientes, por lo que son necesarias modificaciones adicionales, genéticas o epigenéticas, para el desarrollo y evolución de la LLA16.
3Metilación del DNALa metilación del DNA es el mecanismo epigenético más ampliamente estudiado en la LLA4. Esta modificación involucra la adición covalente de grupos metilo al carbono 5 de la citosina del DNA. En mamíferos, la metilación del DNA ocurre principalmente en la secuencia 5’-CpG-3’, conocida como isla CpG17. Los dinucléotidos CpG se encuentran en baja densidad a lo largo de todo el genoma; sin embargo, en las regiones promotoras existe alta densidad de esta secuencia17. Aproximadamente el 70% de las regiones promotoras en mamíferos presentan islas CpG10. La metilación del DNA en regiones promotoras generalmente correlaciona con el silenciamiento en la expresión génica17; por el contrario, la metilación de CpG a lo largo del cuerpo del gen promueve la elongación durante la transcripción8.
Las enzimas responsables de añadir grupos metilo al DNA se conocen como DNA metiltransferasas (DNMTs), y se han descubierto al menos tres en eucariontes18. La DNMT1 se encarga de mantener la metilación del DNA durante la replicación, a través de la metilación de los dinucleótidos CpG recién sintetizados10,18. La DNMT3a y DNMT3b establecen, principalmente, metilación de novo durante la embriogénesis8,10; adicionalmente, reparan errores cometidos por la DNMT1 durante la síntesis del DNA8. Por el contrario, la desmetilación del DNA involucra a la enzima TET2 (ten-eleven-translocation 2), la cual cataliza la conversión de la 5-metil citosina a 5-hidroxi-metil citosina19.
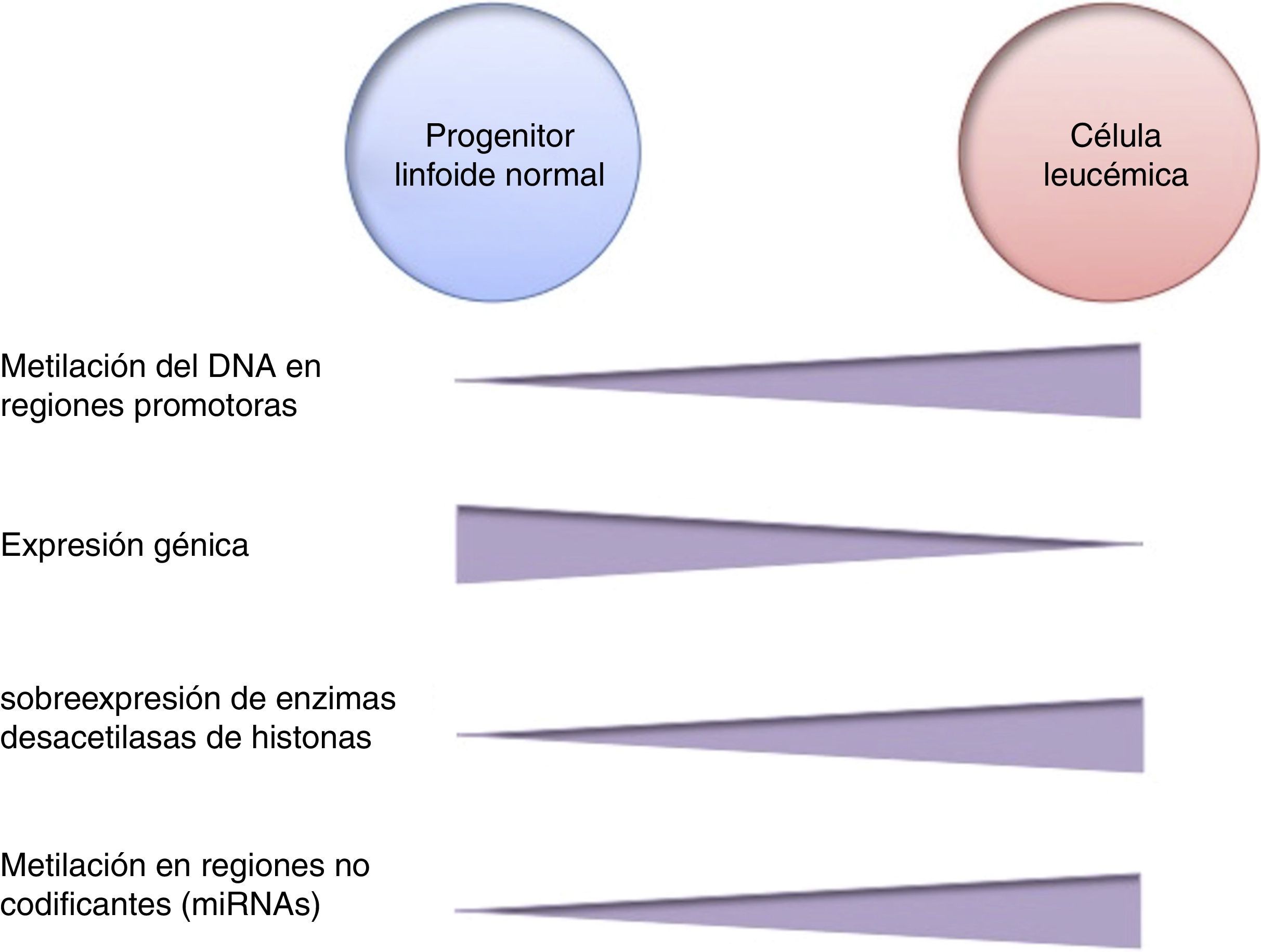
La metilación del DNA es una de las modificaciones epigenéticas más estudiadas en cáncer. En algunos tipos de cáncer, las células tumorales muestran una hipometilación global del DNA que se asocia con inestabilidad cromosómica, reactivación de transposones y pérdida de la impronta genómica4,8. En general, las células cancerosas presentan hipermetilación del DNA en regiones promotoras. Se ha reportado que en el 5-10% de los promotores normalmente desmetilados se presenta metilación anormal en diversos tipos de cáncer10. Esta hipermetilación anormal puede conducir al silenciamiento de genes supresores de tumor, y en consecuencia al proceso carcinogénico18. La hipermetilación anormal en regiones promotoras no solamente afecta la expresión de genes codificantes, sino también la expresión de RNAs no codificantes, los cuales pueden contribuir a la transformación maligna10. La alteración de la metilación del DNA es un evento altamente prevalente en diversos tipos de cáncer, incluyendo a la LLA (fig. 2).

Numerosos estudios han establecido que la alteración en la metilación del DNA tiene un papel fundamental en el desarrollo de LLA de estirpes B y T en adultos y niños. El estudio de alteraciones en la metilación génica en la LLA se ha abordado de dos formas: mediante el estudio dirigido de genes específicos preseleccionados por su importancia en la LLA, y a través de estudios de gran cobertura del genoma en los que el análisis no solo se limita a ciertos genes20. En la mayoría de los casos, el estado de la metilación génica se ha empatado con estudios de expresión génica. En conjunto, estos estudios han revelado metilación y expresión aberrante, principalmente hipermetilación en múltiples genes de manera simultánea, lo que se ha definido como “fenotipo metilador”. Esto conduce al silenciamiento génico, y es una característica común en LLA y en otras leucemias de adultos y niños4,20,21.
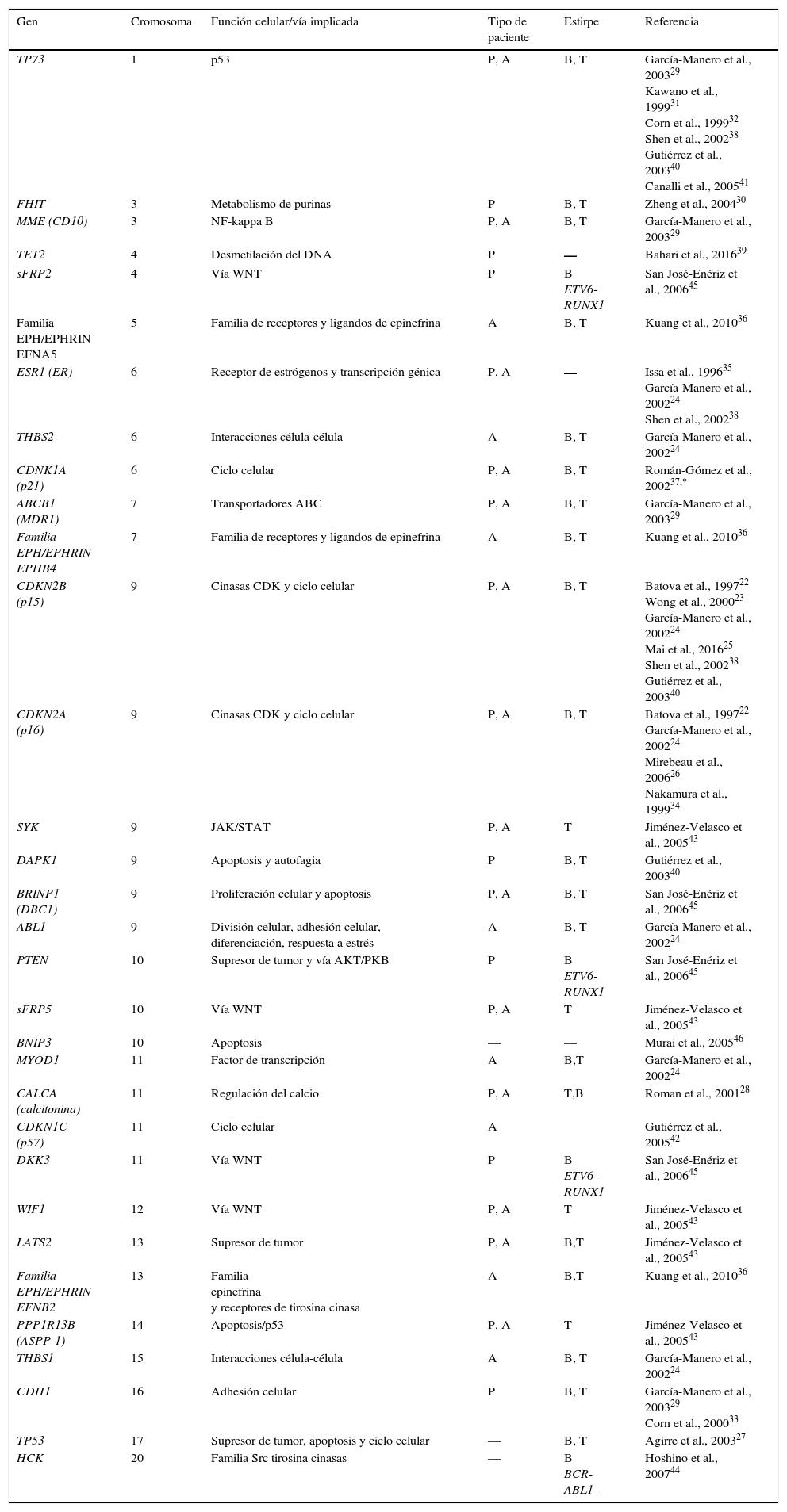
A través de estudios dirigidos, se han encontrado diversos genes con hipermetilación en pacientes con LLA (tabla 1). Algunos de estos son TP73, FHIT, MME (CD10), TET2, sFRP2, EFNA5, ESR1, THBS2, CDKN1A (p21), ABCB1, EPHB4, CDKN2B (p15), CDKN2A (p16), SYK, DAPK1, BRINP1 (DBC1), ABL1, PTEN, sFRP5, BNIP3, MYOD1, CALCA (calcitonina), CDKN1C (p57), DKK3, WIF1, LATS2, EFNB2, PPP1R13B (ASPP1), THBS1, CDH1, TP53 y HCK22–46. La mayoría de estos genes son supresores tumorales y están involucrados en funciones celulares y vías de señalización fundamentales, como las que regulan ciclo celular, apoptosis, respuesta a daño al DNA y expresión génica (tabla 1). A partir de los resultados de los estudios dirigidos, se han obtenido correlaciones entre los patrones de metilación y los subtipos de LLA, así como con el pronóstico de la enfermedad.
Genes hipermetilados en leucemia linfoblástica aguda, identificados a través de estudios de metilación dirigidos
| Gen | Cromosoma | Función celular/vía implicada | Tipo de paciente | Estirpe | Referencia |
|---|---|---|---|---|---|
| TP73 | 1 | p53 | P, A | B, T | García-Manero et al., 200329 Kawano et al., 199931 Corn et al., 199932 Shen et al., 200238 Gutiérrez et al., 200340 Canalli et al., 200541 |
| FHIT | 3 | Metabolismo de purinas | P | B, T | Zheng et al., 200430 |
| MME (CD10) | 3 | NF-kappa B | P, A | B, T | García-Manero et al., 200329 |
| TET2 | 4 | Desmetilación del DNA | P | — | Bahari et al., 201639 |
| sFRP2 | 4 | Vía WNT | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| Familia EPH/EPHRIN EFNA5 | 5 | Familia de receptores y ligandos de epinefrina | A | B, T | Kuang et al., 201036 |
| ESR1 (ER) | 6 | Receptor de estrógenos y transcripción génica | P, A | — | Issa et al., 199635 García-Manero et al., 200224 Shen et al., 200238 |
| THBS2 | 6 | Interacciones célula-célula | A | B, T | García-Manero et al., 200224 |
| CDNK1A (p21) | 6 | Ciclo celular | P, A | B, T | Román-Gómez et al., 200237,* |
| ABCB1 (MDR1) | 7 | Transportadores ABC | P, A | B, T | García-Manero et al., 200329 |
| Familia EPH/EPHRIN EPHB4 | 7 | Familia de receptores y ligandos de epinefrina | A | B, T | Kuang et al., 201036 |
| CDKN2B (p15) | 9 | Cinasas CDK y ciclo celular | P, A | B, T | Batova et al., 199722 Wong et al., 200023 García-Manero et al., 200224 Mai et al., 201625 Shen et al., 200238 Gutiérrez et al., 200340 |
| CDKN2A (p16) | 9 | Cinasas CDK y ciclo celular | P, A | B, T | Batova et al., 199722 García-Manero et al., 200224 Mirebeau et al., 200626 Nakamura et al., 199934 |
| SYK | 9 | JAK/STAT | P, A | T | Jiménez-Velasco et al., 200543 |
| DAPK1 | 9 | Apoptosis y autofagia | P | B, T | Gutiérrez et al., 200340 |
| BRINP1 (DBC1) | 9 | Proliferación celular y apoptosis | P, A | B, T | San José-Enériz et al., 200645 |
| ABL1 | 9 | División celular, adhesión celular, diferenciación, respuesta a estrés | A | B, T | García-Manero et al., 200224 |
| PTEN | 10 | Supresor de tumor y vía AKT/PKB | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| sFRP5 | 10 | Vía WNT | P, A | T | Jiménez-Velasco et al., 200543 |
| BNIP3 | 10 | Apoptosis | — | — | Murai et al., 200546 |
| MYOD1 | 11 | Factor de transcripción | A | B,T | García-Manero et al., 200224 |
| CALCA (calcitonina) | 11 | Regulación del calcio | P, A | T,B | Roman et al., 200128 |
| CDKN1C (p57) | 11 | Ciclo celular | A | Gutiérrez et al., 200542 | |
| DKK3 | 11 | Vía WNT | P | B ETV6-RUNX1 | San José-Enériz et al., 200645 |
| WIF1 | 12 | Vía WNT | P, A | T | Jiménez-Velasco et al., 200543 |
| LATS2 | 13 | Supresor de tumor | P, A | B,T | Jiménez-Velasco et al., 200543 |
| Familia EPH/EPHRIN EFNB2 | 13 | Familia epinefrina y receptores de tirosina cinasa | A | B,T | Kuang et al., 201036 |
| PPP1R13B (ASPP-1) | 14 | Apoptosis/p53 | P, A | T | Jiménez-Velasco et al., 200543 |
| THBS1 | 15 | Interacciones célula-célula | A | B, T | García-Manero et al., 200224 |
| CDH1 | 16 | Adhesión celular | P | B, T | García-Manero et al., 200329 Corn et al., 200033 |
| TP53 | 17 | Supresor de tumor, apoptosis y ciclo celular | — | B, T | Agirre et al., 200327 |
| HCK | 20 | Familia Src tirosina cinasas | — | B BCR-ABL1- | Hoshino et al., 200744 |
P: pediátrico; A: adulto; B: células B; T: células T.
Los estudios de cobertura amplia han incluido un análisis extenso del genoma, así como mayor número de pacientes17,20. Cabe señalar que no en todos los casos se ha encontrado asociación entre los resultados de los estudios dirigidos y los análisis a gran escala, posiblemente por variaciones significativas entre las metodologías y el tamaño de la población de estudio20.
En los estudios de cobertura amplia se han comparado muestras de pacientes LLA-B y líneas celulares leucémicas con tejidos no leucémicos, identificando numerosos cambios en la metilación del DNA, principalmente hipermetilación, en todos los subtipos de LLA-B47,48. Algunos de los estudios se han enfocado en LLA pediátrica o en LLA de adultos y en otros se han analizado ambos grupos. En la mayoría de estos estudios se han incluido pacientes con LLA-B y LLA-T. Taylor y colaboradores estudiaron un número limitado de pacientes, seis niños y cuatro adultos con LLA-B o T, e identificaron 11 genes (DCC, DLC1, DDX51, KCNK2, LRP1B, NKX6-1, NOPE, PCDHGA12, RPIB9, ABCB1, SLCA1) con metilación aberrante (tabla 2) en comparación con células de tejido normal provenientes de sangre periférica y médula ósea. En este estudio no se identificaron diferencias entre la metilación de la LLA de estirpe B y T, excepto en el gen DDX51, el cual no mostró hipermetilación en LLA-T; tampoco se encontraron diferencias entre niños y adultos49.
Perfiles de metilación/expresión alterada en leucemia linfoblástica aguda identificados a través de estudios de cobertura amplia
| Estudio | Población estudiada | Estirpe | Genes con metilación alterada |
|---|---|---|---|
| Taylor et al., 200749 | Adultos: 4 Niños: 6 | B y T | 11 genes hipermetilados: DCC, DLC1, DDX51, KCNK2, LRP1B, NKX6-1, NOPE, PCDHGA12, RPIB9, ABCB1, SLCA1 |
| Kuang et al., 200850 | Líneas celulares mieloides y linfoides Adultos: 61 | B y T | 15 genes hipermetilados: GIPC2, RSPO1, MAGI1, CAST1, ADCY5, HSPA4L, OCLN, EFNA5, MSX2, GFPT2, GNA14, SALL1, MYO5B, ZNF382, MN1 |
| Figueroa et al., 201352 | Niños: 167 | B (137) y T (30) | 85 genes con alteración en la metilación, comunes entre todos los subtipos (73 hipermetilados, 12 hipometilados*): TIE1, LPPR4, ANP32E, *DQX1, *AUP1, *HTRA2, *KCNJ3, GRM7, CEP97, DNAJC19, SENP2, DGKG, CETN3, CAMLG, PCDHA11, PCDHB17, PCDHB11, PCDHB11, PCDHGA5, PCDHGB3, *GNPDA1, PROP1, CMAH, *HIST1H2BJ, *HIST1H2AG, *HIST1H4I, SMAP1, FUT9, ATG5, SLC22A2, HOXA5, HOXA6, CHCHD2, BPGM, AGPAT6, MOS, SDR16C5, WDR67, WDYHV1, ELAVL2, C9orf25, DNAI1, MCART1, TAF3, KIAA1279, H2AFY2, NDUFB8, C10orf26, GPR26, CCDC34, *ELF5, LRRC4C, ZBTB16, SIAE, SPA17, TMTC1, C12orf44, SKA3, MRP63, DHRS12, OXA1L, PABPN1, SLC8A3, SCG5, TGFB1I1, CNOT1, SPNS2, ACACA, TADA2A, LSM12, G6PC3, *GPRC5C, OSBPL1A, INO80C, ZNF560, ZNF582, DDRGK1, ITPA, PTPRT, *OK/SW-cl.69, MAP7D2, GSPT2, PABPC5, MCTS1, MGC16121 |
| Chatterton et al., 201451 | Niños: 69 | B | 370 genes con alteración en la metilación (325 hipermetilados, 45 hipometilados*) comunes entre todos los subtipos: ZFP112, ZFP42, ZNF285, ZNF300, ZNF385D, ZNF677, ZFP37, ZNF560, CXCL1, CXCL3, CXCL5, CXCL6, ADRA1A, ADRA2C, AGTR1, CASR, FPR2, FZD9, GALR1, GPR126, GPR26, GPR37, GPR6, GPR77, GRM1, GRM7, HTR4, LHCGR, OPRM1, P2RY2, *PTAFR, PTGFR, QRFPR, SSTR1, GRIA2, GRIA4, GRIK1, GRIK2, GRIK3, KCNB1, KCNB2, KCNG1, KCNH7, KCNK1, KCNK5, KCNN2, *KCNQ4, TRPC1, TRPC4, TRPM3, SLC19A3, SLC1A1, SLC22A3, SLC24A3, SLC34A2, SLC44A4, SLC5A7, SLC6A1, SLC6A15, SLC6A2, PCDHB15, PCDHB2, PCDHB3, PCDH20, CDH6, CDH8, IL1RN, IL7, *LTA, ACADL, BHMT, CASD1, CYP24A1, CYP7B1, DHCR24, DPYS, DPYSL4, ELOVL4, EPHX2, GALNT11, GCNT2, GDA, LPL, LRAT, ME1, NMNAT3, *NOX3, PDE10A, PDE1C, PDE4B, PDE4C, PLOD2, PNPLA3, PRTFDC1, PTGS2, RGS7, SMPD3, ST6GAL2, UGGT2, CHI3L2, EFEMP1, KL, LYZ, PDZRN3, PLA2G7, POGLUT1, SMPDL3A, *DNTT, RFX6, ABO, ADCY2, *ENPP3, ENTPD3, GNA14, SH3GL2, MACROD2, HS6ST3, BMP7, FGF10, FGF12, GRP, KITLG, NELL1, PTN, NRG1, PEX5L, *VDAC3, CACNA1A, CACNA1D, CFTR, GABRA2, GLRB, GPM6A, HCN1, RYR2, NALCN, *BLK, *HSPB8, PRKD1, MYO3A, MAPK15, PAK7, *DDR1, EPHA5, ERBB4, FGFR2, KDR, NR2F2, PGR, THRB, *ARPP21, *EVL, *FGD2, *KISS1, *S100A2, *SORBS3, *SPRR4, *DEFB129, *MBP, *SFTPB, *WISP1, *WNT5B, *INTS1, *TCL1A, *CLDN15, *GP9, *LY9, *RPH3AL, *XIRP1, *ASB10, *FAM49A, *KLHDC7A, *SH2D4B, *SRBD1, ACTA1, DES, HSPA2, MT2A, NEFM, NEFH, SLPI, VSNL1, LIN7A, TOM1L1, CPLX2, RAB32, ERC2, PDLIM3, STXBP6, GULP1, PARVA, PARVG, NOD2, PREX2, CTTNBP2, RHPN2, PRKCDBP, SPATA18, SPTSSB, ADCYAP1, CBLN4, COL14A1, CYR61, FAM19A4, FAM20A, FSTL5, IGFBP3, LAMA1, LAMA2, MAMDC2, MATN2, NPTX2, RLN1, RLN2, SASH1, SCG3, SEMA3C, SERPINA1, SST, TAC1, TGFBI, TMEM45B, VSTM2A, ZPBP2, ACTL6B, CENPH, KIF6, MIPOL1, MLF1, NOL10, PRDM5, SNRPN, SYCP1, TDRD5, CADM1, CD8B, CDCP1, CLDN11, CNTNAP2, CTNND2, DSCAML1, EPB41L3, FLRT2, LSAMP, MARVELD3, NTM, PCDH8, PKP2, STEAP4, SYN2, THEM4, TPBG, ALS2CR11, C10orf107, C11orf70, C1orf115, C20orf197, C3orf14, C6orf118, C7orf63, CCDC37, CWH43, EFCAB1, FAM83A, FAM84A, FBXO39, FEZF2, FIBIN, LCA5, LPPR1, LRIG3, PHF21B, RBM47, SLC35F3, TMEM171, TMEM74, TSPYL5, TTC22, UNC80, WDR17, XKR6, CTSG, EPHX1, ADAM12, ADAMTS18, ADAMTS5, CPXM2, HP, PAPPA, PCSK1, PCSK2, PRSS12, TLL1, EYA4, PPP2R3A, CDC14B, PTPRO, PTPRZ1, LPPR4, ACTN2, BHLHE22, BNC1, DMRT1, DMRT2, DMRT3, EGR4, FOXA2, FOXG1, GATA4, GCM2, GSC2, HLF, HOXA2, HOXD1, HOXD11, ID4, IRF6, KLF1, *LIMD1, MSX2, MYF5, NACC2, NFE2, NFIB, NKX2-1, NPAS2, ONECUT2, OTX2, OVOL2, PAWR, *POU2AF1, POU4F3, PROX1, PRRX1, SCRT1, SIX1, SNCAIP, SOX1, SOX14, SOX17, SOX9, *TCF3, TLX3, TP73, WWC1, WWTR1, YAP1, ZIC1, EIF5A2, *ANXA9, CXADR, DCC, *ENG, FCGR2A, GPC6, *LRP8, *SCARF1, SFRP1, *HBZ, *SNX8, TF, UPF3A, SORCS3, ATP8A2, *LBP PDPN, SNAP25, SYT9, FAM164A, MOSC2 |
B: células B; T: células T.
Kuang y colaboradores analizaron 23 líneas celulares de origen linfoide y mieloide (MOLT4, Jurkat, Peer, T-ALL1, CEM, J-TAG, B-JAB, RS4, ALL1, Raji, REH, Ramos, K562, BV173, HL60, NB4, THP1, U937, ML1, OCI, HEL, MOLM13, KBM5R) e identificaron 65 genes con metilación alterada. En 15 de estos genes (GIPC2, RSPO1, MAGI1, CAST1, ADCY5, HSPA4L, OCLN, EFNA5, MSX2, GFPT2, GNA14, SALL1, MYO5B, ZNF382 y MN1) se confirmó la hipermetilación al analizar 61 muestras de LLA-B y T de adultos (tabla 2). El análisis ontológico de estos genes reveló enriquecimiento en vías involucradas en la proliferación, crecimiento y diferenciación celular, apoptosis, regulación de la expresión génica, replicación y reparación del DNA, transducción de señales, transporte y metabolismo50.
En el estudio de Chatterton y colaboradores, en el 2014, se asociaron los niveles de expresión y de metilación de 69 niños con LLA-B. Se identificaron 325 genes con hipermetilación y disminución en la expresión génica, así como 45 genes hipometilados y con expresión génica aumentada; estos resultados se compararon con 42 muestras de médula ósea no leucémicas en las que no se detectaron estas modificaciones (tabla 2)51. El análisis ontológico de estos 370 genes mostró una mayor representación de vías moleculares y celulares asociadas con cáncer hematológico, como las señalización e interacción célula-célula, muerte y supervivencia celular, así como desarrollo celular. En particular, se encontró hipermetilación en genes que codifican proteínas involucradas en la señalización del receptor de glutamato, señalización de proteínas G y señalización mediada por adenosín monofosfato cíclico (cAMP); también se encontró represión de proteínas involucradas en el transporte de cationes. La hipermetilación en estos genes, y su consecuente represión, son indicativos de una reducción en el potencial de interacción célula-microambiente, así como en el potencial apoptótico. En este estudio también se identificaron alteraciones en la metilación de genes asociados al proceso leucémico, como las quimiocinas CXCL1, CXCL3, CXCL5 y CXCL6 y las citocinas LTA e IL-7, cuyos genes mostraron hipo e hipermetilación, respectivamente. Adicionalmente, se identificaron 11 genes de cinasas desregulados: BLK, DDR1 y HSPB8, que mostraron hipometilación y sobreexpresión, y EPHA5, ERBB4, FGFRDZ, KDR, MAPKI5, MYO3A, PAK7 y PRKD1, que se encontraron hipermetilados y, en consecuencia, silenciados. También se encontró alteración en factores de la vía de señalización de WNT: se observó hipometilación y sobreexpresión de WNT5B y WISP1, así como hipermetilación y baja expresión de SFRP1, PP2A y SOX. Esta firma de metilación/expresión fue común en todos los subtipos de LLA51. En este mismo estudio también se analizaron firmas de expresión dependientes de subtipos de LLA y se compararon con muestras no leucémicas. Se identificaron 55, 51 y 13 genes con desregulación epigenética asociados con los subtipos ETV6-RUNX1, hiperdiploide y “con otras alteraciones” (este grupo incluyó pacientes con las fusiones TCF3-PBX1, BCR-ABL y translocaciones de MLL), respectivamente (tablas 3-5)51.
Perfil de metilación del subtipo ETV6-RUNX1 de LLA-B pediátrica
| Genes hipermetilados | Referencia |
|---|---|
| NRG1, KCNC3, RAB37, COL7A1, VIPR2, NIPAL1, GREM1, TM6SF1, MTX2, DPP4, DOCK5, SYT17, WWTR1, ITGAX, TMEM144, ASNS, MYO3A, GUCY1B2, CHD5, ITGB5, PAX7, CD44, RAB6B, PI4K2A, ADRA1D, FAM123A, STBD1, OVOL2, RNF207, CRABP1, CDC42BPB, L3MBTL4, KIAA0020, MPZL3, PRICKLE1, ADPRH, PDCD1LG2, SLCO2A1, FXYD6, VAX2, GSX2, SYT12, TMEM67, SLAMF8, CTTN, LPXN | Chatterton et al., 201451 |
| MYO1E, STBD1, OPHN1, HOXD1, WWTR1, SOX3, SNAI3, ZNF462, C4orf31, LRRC55, ST6GALNAC4, C21orf15, ALX1, GUCY2D, FOXA2, ZNF462, SIGLEC9, MTMR11, RIN1, PDE3A, UCN2, KCNN2, NKX2-8, PDK4, NHS, HOXD1, TNFRSF9, NEFL, C20orf165, NEURL2, CTSA, BMP7, SLC35B1, ITIH4, MUSTN1, SLC2A5, MEFV, PLD4, CXCR3, DARC, C9orf79, STBD1, PDK4, UCN2, SRGN, DBX2, C10orf116, AGAP11, ZNF215, BGLAP, PCDHGB4, PCDHGA8, BMP7, PJA2, GPR50, ESX1, KRT80, ZNF804A, TFCP2L1, GNPNAT1, OGG1, ADCY3, CBLN4, CHAT, SLC18A3, PLEKHG4, WNT2, C20orf165, NEURL2, CTSA, GPR123, SYT15, FCN1, BDNF, FBXW5, C8G, C14orf49, EFEMP1, BBC3, LEPREL2, PCDHGB2, PCDHGA5, PCDHGB3, CRYBB3, PTMS, LAG3, FAM190A, EIF2C2, PCDHGA5, PCDHGB3, NEFM, CCBL1, LRRC8A, TUB, TJP1, LIMD1, PPARD, MGC16703, P2RX6, FBXW5, C8G, ZNF296, GEMIN7, GRIA3, NOS3, GABRE, RTDR1, RAB36, ZNF19, MSX2, TSEN2, PRDM11, EPHA5, GPR81, S100A9, EIF6, RPL9, LIAS, SCARF2, CUEDC1, DOK5, HECW1, GPR126, PRKAG3, MARCKSL1, HIST3H2A, HIST3H2BB, SIAE, SPA17, MAGEB6, C14orf49, DAAM2, EIF2C1, TSPAN15, MSRB3, ITGAE, HLF | Figueroa et al., 201352 |
| GRHL3, CTTN | Nordlund et al., 201553 |
| CLIC5, ACVR1C, IGF2BP1, DSC2, POLO | Lee et al., 201555 |
| Genes hipometilados | |
| SLC35B3, EPOR, BEST3, APOBEC3A, MPP7, TMED6, TRIM69, ALPK1, MPP7, CRYBB3 | Chatterton et al., 201451 |
| MIB1, GFAP, TMED6, TPMT, KDM1B, GFAP, E2F6, DRD5, NCKAP1, CSTF3, HTR3C, HTR3C, FUCA1, DNAJC10, B4GALT3, BEST3, CALD1, SIDT1, LRRC4, GNB4, FCHSD2, ABHD3, RAD50, KCNK15, PIK3CG, LRRC4, C22orf24, YWHAH, RRAGA, NACA, VPS13C, SH2D3A, VAV1, C12orf49, RNFT2, ITSN2, TREML1, PRSS1, RAB3GAP1, MRPS15, ATF7, TNK1, ETV7, HLA-DQA1, ERLIN2, ST7L, CAPZA1, TUSC3, BAT2D1, LOC100127888, EID3, TSPYL1, DSE, ISG20, UFC1, ZNF143, SPEM1, C17orf74, C9orf40, MOBKL3, CAB39L, SETDB2, CSTF3, SLC35B3, ATP5J, GABPA, KCTD17, KLHDC5, TOR1AIP2, TOR1AIP1, CIITA, C12orf23, USP6NL, DHRS13, APOBEC3A, CCNI, TOPORS, EPM2AIP1, MLH1, FUT11, TIMM17B, PQBP1, SAA1, THOC7, ATXN7, SIRT2, ZCCHC3, PBXIP1, IRAK3, RAD51L3, FNDC8, TAP1, PSMB9, ISG20, ARHGAP24, PNRC1, DNAJC10, YARS, S100PBP, BTN3A2, RTP4, ELOVL5, DAXX, GAR1, LIMS2, GPR17, SERPINE1, YARS, S100PBP, ZNF644, POMP, RNF113A, NDUFA1, VWA5B2, CTCF, USHBP1, C19orf62, GAB1, C18orf56, TYMS, GABBR1, WDFY3, C4orf12, PRR5L, POLR3B, SPOP, VRK2, HABP4, NUP107, DLGAP2, ZNF566, C1orf59, TMEM127, CIAO1, RECQL4, LRRC14, HORMAD2, PIK3IP1, GLO1, CNR2, FAM71E1, C19orf63, TOR2A, BRWD1, SHE, TDRD10, CDC2L5, C21orf58, PCNT, MRE11A, ANKRD49, ERICH1, HLA-C, CHERP, TRIM9, CMTM2, CCDC62, HSPA8, SMC1B, RIBC2, PDE4DIP, ARHGAP25, USP49, GFI1, NOS1AP, UCP2, CLINT1, GGNBP2, RNF141, EXTL3, EXOSC9, SFT2D3, SEP15, HS2ST1, CACNA1I, ALG9, IKBIP, APAF1, SH2D4B, RAB33B, CLIP1, LOC344967, N4BP2, LOC100127888, LOC492303, ZNF326, FGFR4, HSPB11, LRRC42, HIST1H1C, ARF1, DHRS13, DSC3, SRGAP3, C6orf170, TAF6, CNPY4, MBLAC1, RAB37, CITED2, COG8, NIP7, NDUFA8, MORN5, SRP9, ZNF616, MTA3, FNBP4, GNGT2, ABI3, PELI3, ZNF423, CHDH, IL17RB, PPAT, PAICS, RASAL1, SRGAP3, CST8, ARF6, DIRAS2, LOC100128164, SEC62, MRPS36, FAIM, RAG2, C11orf74, USP50, GPR146, RAB3IL1, ARPP-21, HPS4, SRRD, UBE2J1, KRAS, C14orf142, UBR7, PTPRB, MAN1A2, VPS13B, MLL2, ZFP161, ZC3H7A, CD79B, EBF4, HNRNPK, RMI1, STRBP, OSGEP, APEX1, LOC100128164, SEC62, SMCHD1, SYNGR1, FLJ25006, LOC645851, SENP6, MAP1LC3A, C1orf156, C1orf112, CTHRC1, SCAP, C1QL4, ERCC6, ATPAF2, C17orf39, CALD1, PIK3C3, PAX6, WIPF1, COCH, KCNA3, MTMR10, RAB8B, CANT1, MANBA, POU5F1, RHBDL3, TIAL1, PIAS2, KPNA4 | Figueroa et al., 201352 |
| SESN2, E2F6, RP11-363D14.1, AC131097.3, XIRP1, ZDHHC3, RP11-803B1.2, SIDT1, RP11-71E19.2, APBB2, KIF2A, DUSP1, RP11-274H24.1, TBC1D7, ZNF90P2, CUX1, ERICH1, ERICH1-AS1, ZNF704, FARP1, TMED6, CBFA2T3, IGF2BP1, AXIN2, MIB1, DSC3, EPOR, TCFL5 | Nordlund et al., 201553 |
| SOX11, SPSB1, BEST3, SIDT1, TCFL5, CHL1, FAM19A | Busche et al., 201359 |
Perfil de metilación del subtipo hiperdiploide de LLA-B pediátrica
| Hipermetilados | |
|---|---|
| MYO3A, WWTR1, VIPR2, PID1, FAM20A, SOX11, CRMP1, KL, WWC1, FGFR2, ZNF532, SLIT3, ENPP5, LOX, B3GALNT1, KIAA1456, ADAMTS1, AJAP1, DYNLRB2, AJAP1, MFSD7, DPY19L3, MAGI1, NRG1, PTPRG, FOXL1, FBN2, DSC3, FHIT, DSC3, FBN2, BMPR1B | Chatterton et al., 201451 |
| PPAT, PAICS, KCNF1, HORMAD2, FAIM, APPL1, DUSP15, TTLL9, LCK, CLSTN2, TXNDC11, CSDE1, ASB8, SETBP1, COL4A1, COL4A2, TEAD1, C1orf183, HIST1H1C, OIP5, NUSAP1, SCAP, VPS13B, ARHGAP25, SLC39A1, CREB3L4, RPS13, ABCB10, DSC2, POMP, SLC35B3, PDLIM3, WBP11, C12orf60, ACSM3, SEP15, HS2ST1, SCAP, NPR1, FAT1, INHBE, GLI1, ASAP2, BEST3, LOC145783, TCF12, TFAP2C, CNKSR2, PHLDB2, FBN2, EXTL3, HK2, CDC14B, C11orf30, RELN, TNIP1, HPS4, SRRD, USP11, GFI1, HNRNPK, RMI1, APOBEC3A, CST8, PBX3, ACACA, TADA2A, OSGEP, APEX1, C14orf142, UBR7, LOC100128164, SEC62, SRBD1, HLA-DQA1, PAX6, RAD50, ETV7, CHMP4C, MYOD1, CALD1, CSDA, CNTN2, KPNA4, GFAP | Figueroa et al., 201352 |
| Hipometilados | |
| LOC284837, TNXB, C14orf49, LOC284837, NKG7, KRT36, ENTPD1, APOLD1, LSP1, DBNDD1, CLDN16, C14orf49, SEPT9, CHST15, BIN2, BCL2, BCL9L, CEBPG, VTCN1, CTNND1, KANK2, SLAMF1, LGALS3BP | Chatterton et al., 201451 |
| MX2, C14orf49, C14orf49, ACTR3C, LRRC61, SYNE1, FAM195A, WDR90, ARD1B, BCL3, SYNE1, SERPINA3, SERPINA4, CEP350, DEFA1, DEFA1B, DEFA3, NPBWR2, LRRTM4, ARID3A, C21orf15, ZNF366, MMP26, KRTAP12-1, SERPINA10, PARK2, PACRG, CLCA2, ARID3A, SERPINA4, FAM90A20, DCAF4L2, RAB24, PRELID1, SYN2, SH2D3C, PLEK, TRPT1, NUDT22, GEMIN4, ELP2P, SERPINA10, PNMA3, GPR56, XKR5, NFATC3, BGN, IL13RA1, LOX, TNXB, MX2, GP2, BGN, MAGEC2, AMT, NICN1, KL, KLF6, FAM92A3, ZDHHC21, VTCN1, NRBP2, ABCA12, GPR84, CRIP1, C9orf66, DOCK8, UBXN11, SH3D20, BTBD16, DARC, CYP2F1, ATP2B3, E2F2, B3GAT3, COASY, MLX, MAGEA12, CSAG1, CCT8L2, MMP12, NDUFS8, CLEC4D, C21orf84, KCP, EIF2C2, S100A13, S100A1, C1orf77, CLEC4D, FAM47A, FES, CALML4, COL1A1, PLEKHJ1, NAPRT1, MAGIX, FGD5, C7orf26, IFFO1, NCRNA00095, FBXL19, C9orf116, MRPS2, DCI, C10orf11, SCRIB, PBXIP1, PYGO2, C21orf84, CDK5, SLC4A2, NCRNA00114, STRN4, FKRP, CD300LD, KLC1, CNGB3, DARC, ARHGAP30, NGB, TSEN2, DDAH2, KLHL17, PLEKHN1, RYBP, GEM, FUT11, C9orf79, CRYAA, GP1BB, NOLC1, KRT15, FAM151A, HRCT1, LOC158376, KDM4B, PNMA3, ADAMTSL3, THAP11, TPRXL, TGIF2LY, TGIF2LX, GPR162, COASY, MLX, KIF2B, KRT6C, DCI, KLK6, RFNG, GPS1, C14orf79, ZNF683, PTGDS, LCNL1, SIGLEC7, GPR39, YDJC, CCDC116, SSRP1, P2RX3, FAM189B, LOC642587, FAM195A, WDR90, PCDH12, KIAA1967, ACTRT2, CALML4, TMEM179B, TMEM223, DDAH2, ROBO1, LRRC16B, SLC29A3, S100A3, KRTAP12-1, TMPRSS3, LOC90834, ST3GAL6, SIGLEC9, CDK3, PLA2R1, RFPL3, TLR9, XAF1, POU5F1B, CHPF, TMEM198, IP6K3, CRAT, PPP2R4, HDAC1, BTBD16, MEFV, SIGLEC7, DNAJC4, VEGFB, CRYBB3, CCR7, DCST2, DCST1, STXBP6, ABCB6, C2orf24, FAM134A, ATF6B, FKBPL, RAB37, TMPRSS13, UGT3A2, HAUS7, LY6G6E, LY6G6D, C9orf69, FAM65A, CCDC12, NBEAL2, CCL25, ASGR1, RASSF1, ZMYND10, EIF6, LOC643008, ENTPD3, MEFV, INPP5E, ST6GALNAC4, KRT4, NPW, LIMD1, SLC26A6, SLC2A7, LCN10, LCN6, MAP3K8, SLC25A45, CCDC28B, GRINA, PSAPL1, CTRB2, CSF1R, NMUR1, STOML1, PML, CA5A, PRDX2, RNASEH2A, MUC4, SCARF2, KRT6A, CARD9, C6orf126, C6orf127, LOC401431, ATP6V0E2, PDE11A, C21orf129, NCRNA00112, GPR81, NEK10, SMTN, STXBP6, FCGR2A, GRINA, PCK1, EDNRA, FAM118A, C9orf79, LOC400940, GRM4, CAMKV, DEFA1, DEFA1B, DEFA3, ATF6B, FKBPL, KRT1, XYLT2, LYPD4, DMRTC2, S100A13, S100A1, C9orf79, ATP2B4, PALMD, LOC26102, GPSM1, FAM151A, SCNN1A, CDH5, SIGLEC15, SOX15, BACH1, POLD1, RAB11FIP1, VTCN1, MYEOV, GRIA3, EREG, HSPG2, FGF11, CHRNB1, DENND2D, RUNX1T1, C14orf70, PTH2, CACNG1, PRICKLE2, MUPCDH, SCT, LGALS7B, PTMS, LAG3, C8orf74, CLDN4, GNGT2, ABI3, KCNE1, C8ORFK29, FBXL6, GPR172A, UBE2L6, UCN2, MAGEB6, ZNF19, GRIA3, GSDMD, GNAZ, LOC401431, ATP6V0E2, C19orf59, TRAPPC5, B4GALNT2, NXF4, MEIS3, ATP4A, C9orf116, MRPS2, ARHGAP27, C10orf116, AGAP11, ACD, PARD6A, C16orf48, C16orf86, CPNE7, SH3KBP1, SCARF2, CD79B, SNAI3, LCN10, LCN6, DGCR6L, SH2D3A, VAV1, STBD1, ADAMTSL2, EP400NL, FPGS, WFIKKN2, KLHL5, FXYD3, PIP4K2A, SLC35A2, SMPX, EGLN3, MICALL1, ID2B, S100A9, FTSJ2, NUDT1, ARHGAP24, SHC1, CKS1B, CYP2J2, GANAB, INTS5, MRI1, MFSD10, C4orf10, NAT9, TMEM104, C14orf139, SHARPIN, MAF1, KIAA1875, C19orf59, TRAPPC5, RGS7BP, E2F2, KCNE1, TLR1, DVL2, PHF23, PLCXD2, IRF8, ADM2, MIOX, EPHA2, SSTR2, MYO1A, LZTS1, FLJ90757, BAIAP2, NEDD9, FBXW5, C8G, PNLIPRP2, SPEM1, RXRB, SLC39A7, HSD17B8, DHRS13, SERPINE1, C4orf50, MYH7, MUPCDH, SCT, UBXN6, C17orf61, NLGN2, TCP11, CANT1, FBXO6, SLAMF8, CXCR3, FBXW5, C8G, DYNLT1, SYTL3, HSPG2, MTA2, PARS2, LTA, TNF, C9orf103, LEPREL2, CCDC96, TADA2B, CXorf22, AARSD1, RUNDC1, SOX15, EPHB1, ATP2A1, FASN, PLA2G2E, KIAA1598, C9orf69, APPL2, SP100, FHL2, ITGB2, TRPT1, NUDT22, DNAJC4, MAP6D1, BTBD1, CXCL1, RFTN1, CCL25, GPR126, AOC2, ICT1, CCL17, POU5F1, PSORS1C3, OTOS, PARK2, PACRG, PRKAG3, C1orf56, VPS29, RAD9B, NLRP10, NDN, ANO7, NCLN, PRICKLE3, CTSZ, WFIKKN2, AK1, TMEM37, TUBB2A, KCTD11, TMEM95, KCNK18, CABP1, ANO4, RLN3, IL27RA, SSTR1, EIF6, BRMS1, B3GNT1, ERCC6, DHRS13, C2orf90, VWA3B, GUK1, LOC100240726, ENPP6, HOMER3, NLRP10, COMTD1, CIZ1, DNM1, IL25, CMTM5, CCR6, MARCKSL1, SHARPIN, MAF1, KIAA1875, IL11RA, C1orf231, MMACHC, KCNAB3, TRAPPC1, CNTROB, B3GNT5, ANGPT4, LOC100129637, DEFB124, REM1, PIGZ, FAM96B, CES2, ARMC3, ADRA1D, PTGS1, OR1L4, DAPK2, CMTM2, KCNAB3, TRAPPC1, CNTROB, RAB4A, PRKCH, GPR126, BBC3, CUEDC1, PHLDB1, MAGEB6, HABP4, DLGAP2, TLR2 | Figueroa et al., 201352 |
| S100A16, HNRPLL, DNAJB2, SH3BP5, LARS2, RGS12, RBPJ, DDIT4L, ANKRD33B,RP1-209A6.1, TNXB, CLEC5A, PKIA, EIF2C2, LCN6, C9orf139, FUT7, RP11-393K12.2, MARCH8, ALDH3B2, RAD51L1, SYNE3, SEZ6, MOBKL2A, PLVAP, CLDN14, LOC284837, CELSR1 | Nordlund et al., 201553 |
Perfil de metilación de otros subtipos de LLA-B pediátrica
| Subtipo | Perfil de metilación | Referencia |
|---|---|---|
| MLL translocado, BCR-ABL y TCF3-PBX1 | Genes hipermetilados:WWC1, NELL1, SEMA3C, FGFR2, ARNT2, NRG1, FBN2, KCNC3, SSH3, DSC3, CYB5R2 | Chatterton et al., 201451 |
| Genes hipometilados:CHST12, LSP1 | ||
| MLL translocado | Genes hipermetilados:ZAP70, XYLT2, HLA-B, EDEM1, UBXN11, CD52, C14orf43, HLA-C, MICALL1, C14orf43, PRKCH, TP53I11, GHRL, SNORD10, SNORA67, CD68, MPDU1, ITIH3, CMTM2, PTPRE, MOV10, GUK1, LOC100128164, SEC62, IFITM2, SELO, TOR2A, C3orf37, CD79B, VWA5B2, CD74, C22orf26, LOC150381, ARF1, SORBS3, APPL2, AK1, CCDC102A, GPR114, KPTN, PPP1R14B, PLCB3, KCNK3, LRRC15, ITPRIP, GAB1, PPP1R14B, PLCB3, GP9, CACNB3, FOXN3, FDX1L, AFF3, SNORA77, PRX, CLINT1, UCKL1, UCKL1AS, CAMTA2, FTSJ2, NUDT1, TMEM220, DLGAP2, AMIGO3, GMPPB, KPTN, ANKRD13D, NINJ1, SSTR2, PPP1CA, TBC1D10C, SIK1, SBSN, GAPDHS, PRKCB, IL2RA, KLHDC7A, DEGS1, LIMD1, AK1, CTCF, ZNF267, CCR6, ANGPT4, DENND3, LTB, LST1, MDC1, TUBB, FLJ42393, MCF2L, C1R, HPCAL1, C21orf84, SPAG8, HINT2, SLC19A1, PLA2G4D, NLRP12, GUK1, PRSS27, LSM7, SPPL2B, GADD45B, TMEM220, TMEM204, ANKRD13D, CTDSP2, SH2D4B, STK32C, EFNA3, ANGPT4, SPIB, IRAK3, NDUFB11, RBM10, APP, ARHGAP27, TMEM129, TACC3, SMTN, SPAG8, HINT2, DNAJC10, S100A13, S100A1, NUP107, HIRA, MRPL40, TENC1, OAF, C7orf61, DBNDD1, RPS11, SNORD35B, HSPA8, TAP1, PSMB9, RPAP1, REPIN1, NUDT8, DNTT, STRN4, FKRP, LTA, TNF, TMCO4, HABP4, NCK2, SSTR2, USP50, CXCR5, GYPC, XYLT2, B3GAT3, SIRT2, ARHGAP24, S100A13, S100A1, BCL7C, CTF1, COG8, NIP7, PTPN18, ZFP161,SLC27A2, PPP1R3E, BCL2L2, ITIH3, LOC401431, ATP6V0E2, STK32C, RAB37, MYO7A, GUK1, GJC2, PCNA, CDS2, STK38, HSPB11, LRRC42, STAB1, GP9, NCLN, RGS14, C21orf84, SERPINE1, COG3, CPNE7, BTK, RPL36A, SGPL1, MRPS15, PIK3CG, CRAT, PPP2R4, MDM4, IL3, MLL2, C1orf231, MMACHC, LOC401431, ATP6V0E2, CD22, FFAR1, MAP6D1, BCL7C, CTF1, TMEM127, CIAO1, CNR2, PXK, STX17, ADCK4, ITPKC, CPNE7, CD79B, SHARPIN, MAF1, KIAA1875, IL3, PELI3, SIGLEC15, C3orf42, GHRLOS, NUP205, MAP6D1, PRDM8, DHRS13, FASN, MUPCDH, SCT, LOC100127888, GRINA, RFTN1, IP6K1, AKAP8L, NCRNA00095, FBXL19, MTHFR, CLCN6, C22orf26, LOC150381, PIP4K2A, LOC100128164, SEC62, DAXX, CYFIP2, TNF, LTB, GANAB, INTS5, CLDND2, NKG7, CD37, C12orf49,RNFT2, POU2AF1, SOX15, ZNF366, TMEM127, CIAO1, SFN, LOC100127888, KRTAP5-8, SUV39H2, FAM189B, TIMM17B, PQBP1, ANKS6, TERT, KDM4B, C9orf116, MRPS2, GRINA, ZNRF3, POMP, MAG, MYL4, RHOH, DNAJC10, UGT3A2, BTN3A2, C22orf24, YWHAH, ZNF566, TINAG, ZFHX3, PBXIP1, TGM6, S100A13, S100A1, ACTRT2, ALPPL2, WIPF1, RECQL4, LRRC14, NCKIPSD, R3HCC1, RAB24, PRELID1, SOX15, LY6G6E, LY6G6D, HPS4, SRRD, CANT1, ADM2, MIOX, ATP2B3, SNORA67, CD68, MPDU1, FLJ25006, LOC645851, C21orf129, NCRNA00112, LEPREL2, LTB, LST1, GPER, DHRS13, MUC4, COG3, GNB4, RANBP9, DCUN1D2, TMCO3, RAB33B, ST6GALNAC3, C1QTNF4, STBD1, POLD1, LRRC6, GFAP, LRFN2, MTMR10, FITM1, PSME1, SFT2D1, CBLN3, KHNYN, DAPK3, HBEGF, SIDT1, FUCA1, CSDA, C14orf49, VKORC1, CISH, MAPKAPK3, RAB11FIP1, GFAP, KRTAP12-1, ITSN2, CIZ1, DNM1, TMED6, MAN2A2, MAP1LC3A, SORL1, RAG2, C11orf74, TIMM13, KLHDC5 | Figueroa et al., 201352 |
| CDH3, TBX2, ERCC1, NPR2, DAPK1, CCR6, HRK, LIFR1, DLX3, FHIT | Stumpel et al., 200960; Schafer et al., 201061 | |
| Genes hipometilados:GUCY1A3, JMJD1C, LOC84989, BAZ2B, PRF1, RUNX3, PRF1, MAP7, RHOBTB3, RUNX3, LARGE, SLC25A18, ITGAE, FLT3, NPR1, FAM65B, PTGR1, ACSL1, MEF2C, TRPV3, SKA1, CNKSR2, H1F0, GCAT, F3, MTHFD2L, TLR7, ARRDC4, DAPK1, PSMB5, TMEM41B, LOC100303728, SLC25A5, ZNF367, FLT3, LOH12CR2, LOH12CR1, PHLPP1, SERPINB13, OR6V1, PRDM11, CD3D, CD3G, SENP6, TRAK2, STRADB, ASTN2, TRIM32, SETBP1, IGF2BP2, TFCP2L1, CENPF, MPP5, C7orf13, RNF32, PSMB5, C7orf13, RNF32, CD180, PARP8, RBKS, LOC100302650, BRE, ARID1B, C6orf122, C4orf19, MGC16703, P2RX6, OLFML3, IQSEC1, MYST2, KRTAP10-6, PDPN, NID1, CPD, LCE3A, SSR1, CDKL3, UBE2B, PAPOLA, ANKRD27, RGS9BP, HTR7, DES, FAM5B, CDC14B, GPR153, SPOCK2, ITGAE, LECT2, SERPINB8, ZAR1L, BRCA2, CAMTA1, C9orf100, ALDH4A1, C17orf64, KRT81, LILRB4, MRPS25, ASB8, SLC25A38, TRAS, MYOD1, DKK3, PPP1R3B, BDNF, CCBL1, LRRC8A, RASGRF2, GRIK5, KCNJ10, FAIM, MTMR11, PEA15, SBF2, HECW1, WDYHV1, PHLDB2, GINS3, PDE4DIP, KDM5B, ZNF256, PHLDB1. | Figueroa et al., 201352 | |
| ZSCAN18, ZNF256, ZNF329, ZNF544, ZNF681 | Nordlund et al., 201353 | |
| BCR-ABL | Genes hipermetilados:KIAA1949, NRM, MUPCDH, SCT, ATP2A1, FASN, DPM2, PGLS, MYEOV, DUSP5, MARCKSL1, ANKRD27, RGS9BP, ADCK5, FOXH1, C11orf16, DIRAS1, MRPL24, DCST2, DCST1, CCDC71, INPP5E, SLC22A20, GPR146, PLEKHM2, PROC, DIRAS1, ALDH4A1, WFIKKN2, IL11, LIMS2, GPR17, GRINA, LILRB4, C19orf59, TRAPPC5, CD38, LOC100129637, PRPH, LOXL4, KLHL24, ARHGAP27, GPR146, SSR1, C19orf59, TRAPPC5, C17orf61, NLGN2, C9orf116, MRPS2, DLG4, ACADVL, TLR9, STOML1, PML, KLF13, KCNK17, KLC1, ARHGAP30, C8ORFK29, FBXL6, GPR172A, DLG4, ACADVL, TAF6, CNPY4, MBLAC1, C9orf69, AURKC, PCOLCE, PTMS, LAG3, DNM1P35, GRINA, BSCL2, GNG3, CCL25, TEX19, CDH5, TLR9, CDK5, SLC4A2, GUK1, C9orf116, MRPS2, S100A13, S100A1, FXYD2, GPR153, LOC100129637, FLJ90757, BAIAP2, IP6K3, KLF6, LPAR2, UCN2, KCTD11,TMEM95, TFR2, C14orf70, CARHSP1, KLF13, CCDC96, TADA2B, LIMS2, GPR17, COL1A1, ARID3A, SHARPIN,MAF1, KIAA1875, SIRT2, WFIKKN2, ZNF296, GEMIN7, RFNG, GPS1, SLC26A8, MAPK14, FAM176B, CCBL1, LRRC8A, KCNK17, KCNAB3, TRAPPC1, CNTROB, MAGIX, PCDH12, SHARPIN, MAF1, KIAA1875, C16orf59, C9orf69, SP100, LACE1, C17orf64, CSF1R, MUC4, C1QL4, KCNAB3, TRAPPC1, CNTROB, GRB7, CLDN4, CARD9, ATP1A3, C1orf220, GUK1, FAM109B, C22orf32, CLTB, HSF2BP, RRP1B, GABBR1, B3GNT5, ITIH3, LOC26102, GPSM1, UBXN6, SH3D20, SIGLEC7, EDC4, CCR7, ACTRT2, CTXN1, RAB3IL1, DNAJC4, VEGFB, ADM2, MIOX, TUBB2A, SSX2IP, C16orf81, LIMS2, GPR17, ZNF48, PKNOX2, GRASP, PECR, TMEM169, NTN5, C9orf103, CACNA1I, BTG2, ADRA1D, DHRS13, THEM4, MT1L, PHLDB2, PDE4DIP, IL25, CMTM5, LIF, KLHDC5, PITPNM3, GALP, FAM109B, C22orf32, PIGZ, PRDM11, CNTN2, MIB1, BSN, MOCOS, DRD5, COL1A2, KIAA1257, LYPD4, DMRTC2, POU5F1, RASL11B, GFAP | Figueroa et al., 201352 |
| Genes hipometilados:GBA2, RGP1, DPP3, C11orf30, ECHDC1, ZEB2, INHBE, GLI1, INTU, WBP11, C12orf60, STARD3NL, C21orf59, FGF6, ITGA2, FBXW7, OIP5, NUSAP1, C6orf47, BAT4, CSNK2B, RPS13, RPL4, SNORD18C, SNORD18B, SNORD16, SNORD18A, ZWILCH, CCDC115, IMP4, POLR1E, C22orf26, LOC150381, ANG, RNASE4, SRBD1, GIMAP5, C1orf183, SLC25A28, TRIB3, CTNNA2, STBD1, C22orf26, LOC150381, IQGAP2, RHBDD3, EWSR1, CSDA, C3orf42, GHRLOS, CYP1B1, GPR6, MFI2, TTC23L, OGFRL1, LRRTM1, CCDC70, C5orf43, NKX2-1, ADAMTS12, NDUFC1, NARG1, TUBA4A, TUBA4B, CLIC2, TMEFF2, KIAA0087, UGGT2, CACNA1B, PRPF8, PJA2, CXCL12, SLC20A1, ACSBG1, CA6, PDZRN3, RTDR1, RAB36, ROBO4, BRDT, PROX1, COL14A1, FLJ14107, ATOH1, NLGN4X, SLC35A1, LRIG3, LANCL1, CPS1, HOXD1, KCNN2, PCNA, CDS2, IL2RA, ZNF264, PCDHGB7, PCDHGA11, GAB1, FGF5, SALL3, CISH, MAPKAPK3, C15orf24, PGBD4, SFN, RIC3, FAM190A, LIMD1, MTMR11, EPHA5, IRX2,C5orf38, ZNF804A, HIPK2, TBXAS1, TINAG, BPNT1, IARS2, MSX2, GLIPR1L2, PRDM8, BDNF, MTERFD2, HOXD1, LRFN5, CDCA5, ZFPL1, MAL2, CDH8, CD22, PCDHB18, PCDHB19P, KCNK13, PKD2L2, GZF1, PDPN, VKORC1, DAXX, FOXQ1, TNFRSF9, MRPL1, TRIM13, UCHL5, TROVE2, EFR3A, SLC39A1, CREB3L4, WIPF1, XYLT2, CPVL, RAG2, C11orf74, PCDHB7, PCDHB8, BCL2L13, METTL8, DCAF17, LRP11, CHMP4C | Figueroa et al., 201352 | |
| TCF3-PBX1 | Genes hipermetilados:FNBP4, CLDND2, NKG7, ARPP-21, SOCS2, NCK2, ARHGEF6, GNA13, TLR1, TMEM156, CD79B, HABP4, ISG20, FOXM1, C12orf32, LAIR1, HECA, CST4, STX17, NFKBIE, TMEM151B, PIGV, MLKL, FDXACB1, C11orf1, ASGR2, EFHC2, GNGT2, ABI3, USHBP1, C19orf62, ZNF25, OAS3, CYSLTR1, GAB1, GNGT2, ABI3, SORBS3, KRAS, BEST1, ARAP3, POLD1, HSPB11, LRRC42, RAB37, ITPRIP, MTA3, KIAA2013, SERINC5, ARHGAP24, KLHL5, CCL25, IL2RA, B3GNT9, TRADD, FBXL8, UBE2L6, KLHL21, GABBR1, OLFML3, NGF, C5orf56, PCNA, CDS2, SSRP1, P2RX3, TMEM127, CIAO1, STAB1, ETFA, TSFM, LOC100129066, ZNF683, ARHGAP24, HSD17B4, CPNE8, NUP107, UGT3A2, FBXO6, CLEC4D, VRK2, C11orf75, PDK1, CCL17, PSTPIP1, CEP350, IKBIP, APAF1, VWA3B, HGF, TMEM217, TBC1D22B, C17orf64, RNF113A, NDUFA1, HELZ, BTG2, SMARCB1, EIF4E3, GPR27, MEF2C, DUSP4, CTDSP2, NUDT5, CDC123, ATP5G2, ST8SIA1, PRDM8, CTCF, SMTN, CMTM2, DAXX, KLHL24, CACNA1I, NCLN, PTPLAD2, ERCC6, HBEGF, PRDX1, FCGR2A, CLU, TNFAIP8, LTB, LST1, FOXN3, MCM3AP, C21orf57, SMC1B, RIBC2, BTG2, SMC6, GEN1, DAZAP2, SYNGR1, CDC2L5, LMAN2, UTP6, C22orf24, YWHAH, SIGLEC15, LOC646999, STRA6, NOLC1, NCAPH, SENP6, ZNF423, NCKIPSD, PPP1R15B, SERPINE1, KPTN, FAM60A, FLJ13224, LDLRAD3, LCN10, LCN6, SLC38A5, FTSJ1, SPOCD1, FPGS, ANKS6, WIPF1, MTMR11, MRPS15, NOLC1, C20orf197, ZNF652, DPY19L3, ATP2B4, SRGAP3, EN1, ZNF41, CYBRD1, RANBP9, LRP11, LTB, LST1 | Figueroa et al., 201352 |
| Genes hipometilados:ZNF512, CRYM, WAPAL, SLAMF1, MYBPH, EXTL3, FAM109B, C22orf32, LHCGR, AKR1B1, SURF1, SURF2, PLXNC1, WDR87, SIPA1L3, NOL11, EXTL3, SDF4, B3GALT6, DEF6, NCRNA00176, LIPC, NFKB1, ASB8, TERT, NSUN4, LOC100129534, ADIPOQ, PHYHD1, BLK, LPAR2, FAM109B, C22orf32, GJA4, ADIPOQ, PDCD1, MND1, RASL11B, RNF186, PSMC3IP, MRPL28, ACTL9, TNS4, LAT, FAM109B, C22orf32, TEKT2, ADPRHL2, NAGA, FAM109B, C22orf32, PLD4, TEKT2, ADPRHL2, C16orf11, GLYCTK, NGF, PIP4K2B, ACOXL, MAST4, AURKC, SOST, MAP6D1, POLM, MSRB3, VAMP2, SLC39A1, CREB3L4, IDH2, NAT1, SSR1, ST6GAL2, FXYD2, ST6GALNAC3, CDCA3, USP5, PHACTR3, FFAR1, SDF4, B3GALT6, GDPD5, ARL4C, ATXN7L2, FAT1, GLYCTK, TRIM67, CTBP1, C4orf42, ARL4C, HOXD1, TSPAN3, TBX6, YPEL3, B3GNT7, MUPCDH, SCT, HIST1H1C, ITIH3, C12orf53, SOX21, SALL3, HECW1, PEA15, MNAT1, WNT2, PPARD, C16orf81, LILRB4, WNT2, C9orf100, SOX9, NCRNA00176, TFAP2C, C16orf81, GPR123, HOXD1, NXPH2, USP11, LTA, TNF, SCAP | Figueroa et al., 201352 | |
| CRLF2 rearreglado | Genes hipermetilados:STXBP5, SLC2A8, GFRA1, DSC3, C9orf150, LCE3A, CHRDL1, C15orf24, PGBD4, HOXB2, ST6GAL2, NOVA1, NOL4, SLITRK1, TMSB15A, PRPF19, LRRC57, HAUS2, C18orf34, EPHA5, DSC2, RBPMS, TUBGCP3, AQP4, C18orf16, GABRQ, TMTC1, AADAT, ZIC5, MYEOV, ASXL1, NEUROG1, EPHA5, SIX6, TMED3 | Figueroa et al., 201352 |
| Genes hipometilados: HIPO, LYSMD4, SRBD1, PYGM, CASQ1, KCTD3, ANO7, WIPF1, CA6, GP1BB, NLRX1, LCN10, LCN6, SLC2A5, S100A13, S100A1 | ||
| ERG alterado | Genes hipermetilados:CDKN2C, CYSLTR1, TMEM156, PIGV, RFTN1, SOCS2, MTHFS, MLKL, SERINC5,CD48, FLJ14107, ENAH, TNF, LTB, DAXX, RFTN1, TNFAIP8, TUBA4A, TUBA4B, HIBCH, NUBPL, C9orf25, DNAI1, STIM2, CHMP1B, PEX16, GYLTL1B, TLR1, CH25H, DEGS1, C8orf56, BAALC, XPO5, POLH, CXCR3, MED14, DDX3X, DENND3, ZNRF3, PELI3, PIK3IP1, HELZ, TGM6, GAB1, RBP1, C1QL4, ATP2B4, PRR5L, SLC27A2, ATP10A, LTB, LST1, STIM2, TMEM178, BNIP3L, TNFAIP8, CEP350, ZMYND8, UBE2J1, CLINT1, EXOSC9, MTMR10, FPGS, CCNI, CDKN1A, UBXN11, FAM120AOS,FAM120A, SYNGR1, ITGB2, RNF141, CD3D, CD3G, SRGAP3, CHRNA2, ZNF490, ZNF791, TP53BP1, WBP11,C12orf60, WDFY3, C4orf12, CARD8, GGNBP2, IKBKE, UTP20, RAPGEF1, HSPA8, PPP1R15B, SENP6, ST6GALNAC3, CD48, KDM4B, PPID, ERO1LB, SH2D3A, VAV1, KCTD17, ZNF212, ARHGAP24, RANBP10, TSNAXIP1, CYTIP, LTB, LST1, LOC729234, PKNOX2, C8orf56, BAALC, SYNE1, DYNLT1, SYTL3, TRIM13, UBR5, IGFBP7, ERAP2, CDCA5, ZFPL1, PARS2, MEF2C, PIP4K2A, NLRP12, ANKFY1, ST3GAL6, PJA2, UGT3A2, DHRS13, FUCA1, CENPJ, RGMA, RAB39, LRRC8D, RANBP9, AIF1, ITPRIP, TAF3, ERCC6, MRPS15, SRGAP3, CDCA3, USP5, NGRN, PIP4K2A, L1CAM, SERPINE1, EFHD2, FXYD5, NUP107, NARG1L, MAP3K8, C14orf33, KTN1, RPRML, RNF113A, NDUFA1, ADAMTS5, MDM4, TLR7, EGLN3, HYAL3, NAT6, HYAL1, NOVA1, DCTN2, KIF5A, TRIM9, ST6GALNAC1, C1orf127, WIPF1, LRRC57, HAUS2, CYBRD1, SLAMF1, ACOXL, PBK, ECM1, RASL11B, TMEM37, GRPEL2, ACSL3, ICAM3, C1orf156, C1orf112, PXT1, KCTD20, STXBP6, TBCCD1, DNAJB11, SIDT1, FGFR4, SLC16A9, CAB39L, SETDB2, ERICH1, DTX3, SSTR2, CBX6, VRK2, C14orf48, EIF4E3, GPR27, SMCHD1, XYLT2, TSEN2, CEP70, STXBP6, USHBP1, C19orf62, ST6GALNAC2, ADAP2, GLRA2, WFDC2, NOLC1, GFI1, CD180, NP, SLC39A13, IRAK3, SPRYD5, C9orf79, DNAJC9, MRPS16, LYPD5, ZNF283, TMEM217, TBC1D22B, ENTPD3, HSPG2, CCDC85A, STK38, CNR2, KIR3DX1, IL8RA, CECR2, MYL12A, MYST2, KCNAB1, WDR47, PYY, NAGS, GNAZ, DEFA1, DEFA1B, DEFA3, C17orf64, RPL17, SNORD58C, U58, SNORD58A, SNORD58B, ESPL1, PXK, CARD14, HNF4A, C9orf79, PCDHB2, CD33, DGKG, S100A13, S100A1, FIBIN, ATP5G2, RAB11FIP1, GREB1L, GJA4, PRICKLE2, RAG2, C11orf74, ZCCHC9, ZNF683, CXXC5, HNMT, C12orf23, ACSBG1, CLU, BPNT1, IARS2, CCR7, ALX3, GABRR1, TFB2M, C1orf71, DDX19A, CACNA1S, THYN1, ACAD8, PTH2, KIF12, GNGT2, ABI3, CAMK2B, SEMA4A, ADARB2, MC4R, DNAJC10, TMPRSS13, C7orf43, DOCK3, DCK, NBLA00301, STBD1, USP2, CDR2, SSRP1,P2RX3, IKBIP, APAF1, PNPT1, DCAF5, ZNF423, PLEKHH3, CCR10, CNTNAP1, LEMD1, PCDHB7, PCDHB8, MLL2, XPNPEP2, DYNC2H1, CPOX, PIAS2, GMNN, HMP19, SEL1L3, PTPRB, CRYBB3, C14orf115, NELL1, ITIH5, RRAGA, LRAT, DEFB124, REM1, C15orf24, PGBD4, B4GALT3, RHBDL3, SRBD1, HTR7, FAM193B, GNB1L, C22orf29, AFAP1, BRDT, MRPL1, DIAPH2, PRICKLE3, ACTRT1, TNFRSF9, SSTR2, HNF4A, SOCS3, SLC39A1, CREB3L4, KCNAB3, TRAPPC1, CNTROB, FOXO4, NOV, NOLC1, DNAJB1, C9orf79, LOC400940, TMPRSS2, GPR84, ZNF366, SFT2D3, TUSC3, VPS72, DCBLD1, AMMECR1L, SLC2A14, PRR18, VPS24, DNAJC10, EXOC8, C1orf124, MAL2, TPRXL, YARS, S100PBP, PO | Figueroa et al., 201352 |
| Genes hipometilados:A2LD1, SAP18, BTN2A1, OSBPL11, HN1, AARS2, ADI1, OBFC2B, SLC39A5, ZNF295, PARVB, MIA, RAB4B, ZNHIT2, FAU, MRPL49, TULP4, C7orf65, TMIE, PSMA2, MRPL32, SPINK4, ZNF296, GEMIN7, PEX11A, WDR93, RASSF2, PNRC1, SLC39A11, GIT2, ANKRD13A, DIXDC1, VPS4B, PARVB, CHRNA1, KRTAP5-8, ADAMTS9, CNTN2, C14orf139, EPS8, PCNX, LDOC1L, CCDC109A, PRMT10, CCNT1, NSD1, WDFY4, DCP1B, GCHFR, C12orf36, RFC4, TFF3, GPR81, ZNHIT6, C19orf23, C19orf24, PIK3R1, C16orf11, NHLRC4, PIGQ, C10orf118, TDRD1, XPNPEP2, SECISBP2, GLYCTK, CUL7, MRPL2, KLC4, GLG1, C7orf25, GPR153, EIF1AD, BANF1, PPL, TMEM127, CIAO1, GPR146, CAND2, DIXDC1, UCKL1, UCKL1AS, SNHG7, SNORA43, SNORA17, RFC4, NME3, MRPS34, EME2, ING1, USP12, HNRNPR, RGN, NHLRC4, PIGQ, TSKU, GRB2, CST8, SCGB3A1, ZNF513, ADCY1, LRRN1, C3orf27, EXOSC10, LFNG, XYLT1, C14orf126, GPR44, CRBN, C16orf81, GLIPR1L2, LTA4H, SPIB, CCNJ, GPR146, LOC643008, GLIPR1L2, BACH2, CCDC109A, GPR155, ZC3H18, ELP3, ALPPL2, CRBN, ADAMTS3, POLM, MRPL28, HEATR5B, CCDC75, GPER, CUL7, MRPL2, KLC4, CHRNA1, C14orf139, SREBF1, MRPL55, KCNC3, BTG2, MTSS1L, OR2Z1, LOC100303728, SLC25A5, CACNA1I, IFI30, GRINA, CRHBP, RNASE7, CD81, SURF1, SURF2, PTOV1, TGFBR2, TEKT2, ADPRHL2, ADAMTS3, IGSF21, SOHLH1, KCNT1, RPL9, LIAS, SHARPIN, MAF1, KIAA1875, GRINA, C20orf165, NEURL2, CTSA, SLC20A2, C8orf40, SNW1, C14orf178, RHBDF1, MPG, LOC26102, GPSM1, EIF4G2, RHBDF1, MPG, TPI1, GLYCTK, ATP2A3, ARHGEF4, TBC1D13, CD38, MS4A10, LOC100129637, SOX15, LIF, C19orf23, CIRBP, C19orf24, ABCA2, C9orf139, CCDC96, TADA2B, BSCL2, GNG3, NELL1, RGS9, GPR148, GANAB, INTS5, HIST1H1C, BMP7, LOC100129637, CGREF1, ABHD1, PCP2, STXBP2, LOC643008, CIZ1, DNM1, GPER, TSKU, TRIM67, GFRA1, FLJ90757, BAIAP2, FTSJ2, NUDT1, DDX50, FOXJ2, SLCO3A1, LIN7A, GATM, RAMP1, COG8, NIP7, NFS1, ROMO1, VPS37C, FAM53B, G0S2, SHANK1, SPATA24, C19orf23, CIRBP, ZNF566, FAM83H, IMMP2L, LRRC3, TSTA3, HLF, SDF4, B3GALT6, LHCGR, C16orf42, GNPTG, SLC2A8, HS6ST1, C20orf165, NEURL2, CTSA, SFT2D1, CSMD1, RAD54L, PITRM1, MUPCDH, SCT, TIGD3, VAT1L, TOMM40, CCDC42, WARS, WDR25, GRIK5, CYP2U1, ODF3L2, HMP19, KLHL22, ARID3B, MT3, SOX15, DMRT3, REPIN1, MTA2, ATPAF2, C17orf39, SHARPIN, MAF1, KIAA1875, SQLE, CHMP4C, SYT15, MYH6, SNTB1, MRPL24, GPR123, GATA6, FBXW5, C8G, EPHA5, PRELID2, IVNS1ABP, KRT80, SNAI2, RBM3, VWDE, NUDT10, ZMYM6, MRPS30, TIMM13, ZNF532, SYVN1, NKX2-8, KIAA1024, FAM81A, KCNN2, MUC1, THYN1, ACAD8, LOC100130557, NFYC, ALOX5, CCDC58, FAM162A |
Figueroa et al. consideraron en su estudio pacientes con LLA con ambos inmunofenotipos; incluyeron 137 niños con LLA-B y 30 con LLA-T, e identificaron una firma de 85 genes con alteración en la metilación, que correlacionó con el análisis de expresión (tabla 2). La firma identificada fue común entre todos los subtipos de LLA (B y T) e incluyó a los genes TIE1, MOS, CAMLG, GPRC5C, involucrados en señalización; MCTS1 y DGKG, que participan en regulación del ciclo celular y proliferación; PABPN1 y PABPC5, que participan en metabolismo de RNA; PROP1, TAF3, H2AFY2, ELF5, ZBTB16, CNOT1 y TADA2A, involucrados en regulación transcripcional, así como los genes homeóticos HOXA5 y HOXA652. Adicionalmente, en este estudio se determinó que existen firmas de metilación y expresión características de cada subtipo de LLA-B pediátrica. Se identificaron siete grupos de LLA-B independientes que presentaron una firma de metilación/expresión característica, dependiente del rearreglo molecular presente en cada grupo de pacientes: con CRLF2 rearreglado, ETV6-RUNX1, hiperdiploidía alta, MLL translocado, ERG alterado, BCR-ABL1 y TCF3-PBX1 (tablas 3-5).
El estudio de Norlund y colaboradores, que incluyó un mayor número de pacientes con LLA pediátrica (663 niños con LLA-B y 101 niños con LLA-T), confirmó que cada subtipo de LLA presenta una firma de metilación específica. Con base en este estudio, se propuso que las firmas de metilación podrían ser útiles para predecir el subtipo genético de la LLA pediátrica; esta propuesta sería aplicable en los casos en los que se dificulte identificar una alteración cromosómica o molecular que contribuya a la clasificación de la leucemia53. Sin embargo, a pesar de que se han encontrado firmas de metilación que podrían determinar el subtipo de LLA y se han propuesto perfiles epigenéticos que permitirían predecir riesgo a recaída47,54, aún no se conoce un perfil epigenético que certeramente sea capaz de clasificar y estratificar a los pacientes con LLA48.
3.2Alteraciones en la metilación génica en LLA de acuerdo con el subtipo específico de rearreglo genéticoAdemás de las alteraciones en la metilación del DNA, comunes en todos los subtipos de LLA-B, existen modificaciones exclusivas de cada subtipo genético48,51,55. Recientemente, se analizaron los perfiles de metilación en conjunto con perfiles de expresión génica de pacientes con la fusión ETV6-RUNX1; se identificaron 55 genes con alteración en la metilación (tabla 3)51. Entre estos se identificó al gen EPOR, el cual se encontró sobreexpresado, y su promotor, hipometilado. La fusión ETV6-RUNX1 se une y activa la transcripción del receptor de eritropoyetina EPOR, el cual contribuye al desarrollo de leucemia, activando la proliferación y supervivencia celular a través de la vía JAK2-STAT556. La hipometilación en EPOR favorece que su promotor esté más permisivo a la unión y activación por ETV6-RUNX1.
En el subtipo ETV6-RUNX1 también se encontró hipermetilación y baja expresión en el gen de la asparagina sintetasa, ASNS, que sensibiliza a las células al tratamiento con L-asparaginasa. Esto explica, en parte, el hecho de que los pacientes con ETV6-RUNX1 presenten buena respuesta a tratamiento57,58. Otros genes con alteración en la metilación y expresión observados en pacientes ETV6-RUNX1 positivos incluyen a CLIC5, ACVR1C, IGF2BP1, DSC2, PCLO, SOX11, SPSB1, BEST3, SIDT1, TCFL5, CHL1 y FAM19A (tabla 3)55,59. Adicionalmente, se ha reportado que la alteración en la metilación de ERHV-3, DMNBP, KCNA3, PAG1 y C11orf52 se encuentra asociada con el riesgo de recaída en pacientes con ETV6-RUNX1, por lo que se han propuesto como marcadores de mal pronóstico47.
En pacientes con hiperdiploidía también se han encontrado alteraciones en la metilación y expresión génicas exclusivas de este subtipo. Los estudios más recientes resaltan la hipermetilación y baja expresión de los genes supresores de tumor FHIT, PTPRG y DDIT4L47,51. Adicionalmente, se encontró hipometilación de los genes proapoptóticos BCL2 y BCL9L, los cuales se han reportado alterados en diversos tumores de células B (tabla 4)51. Por otro lado, el subtipo de LLA positivo para rearreglos en el gen MLL también presenta un perfil característico de alteración en la metilación (tabla 5). En LLA con rearreglos en MLL, se ha reportado la hipermetilación de los genes CDH3, TBX2, ERCC1, NPR2, DAPK1, CCR6, HRK, LIFR1, DLX3 y FHIT60,61. La hipometilación de los genes ZSCAN18, ZNF256, ZNF329, ZNF544 y ZNF681 se ha asociado con un incremento en el riesgo de recaída en pacientes con alteraciones en el gen MLL47. En particular, se ha observado que las LLA con diferentes translocaciones en MLL presentan un perfil de metilación distinto60.
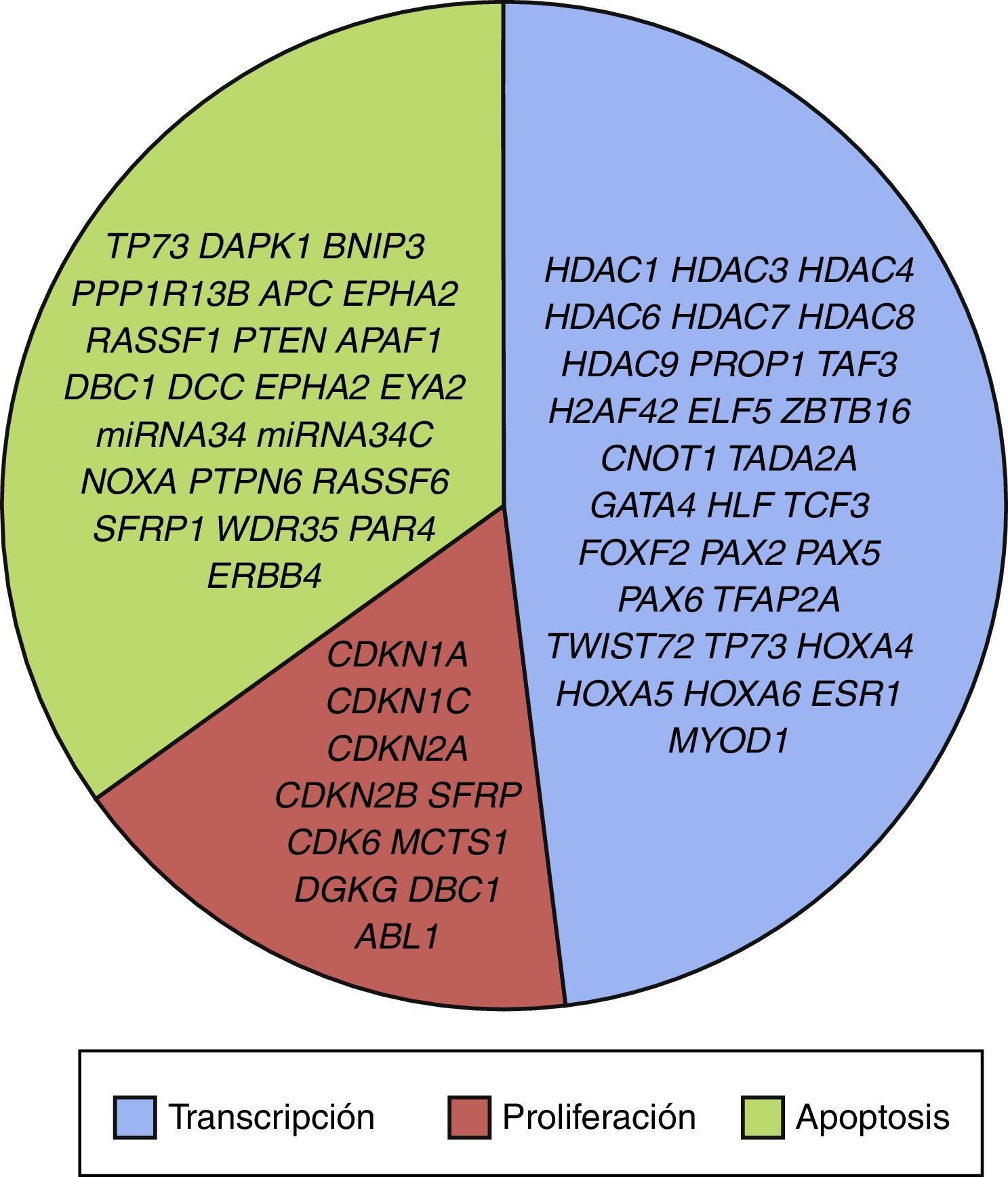
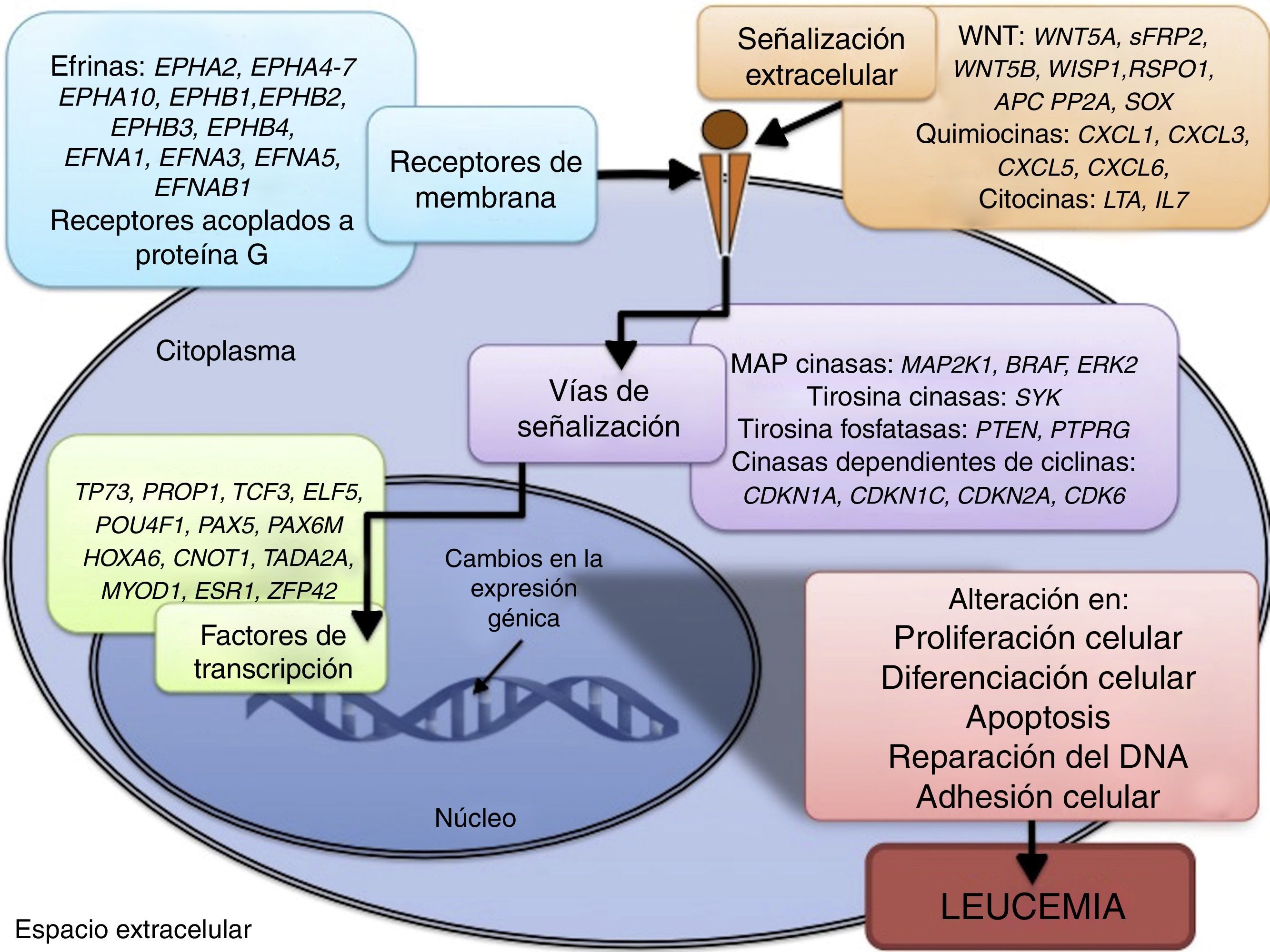
3.3Principales procesos celulares y vías de señalización afectadas por alteración en la metilación génica en LLALos procesos celulares con mayor representación de genes con metilación aberrante en LLA incluyen la regulación del ciclo celular, apoptosis, regulación transcripcional y adhesión celular (fig. 3). Algunos ejemplos de genes con alteración en la metilación en LLA que participan en estas funciones celulares son CDKN1A, CDKN1C, CDKN2A, CDKN2B, SFRP, CDK6, que regulan la proliferación y ciclo celular62–66; GATA4, HLF, TCF3 FOXF2, PAX2, PAX5, PAX6, TFAP2A, TWIST72, TP73, HOXA4, HOXA5 y HOXA6, que participan en la transcripción génica51,62,67–70; DAPK1, APC, EPHA2, RASSF1, PTEN, APAF1, DBC1, DCC, EPHA2, EYA2, miRNA34, miRNA34C, NOXA, ASPP1, PTPN6, RASSF6, SFRP1, WDR35, PAR4, ERBB4, implicados en apoptosis4,20,36,51,71–73. La alteración en la metilación de estos genes confiere a las células leucémicas ventajas de supervivencia, previniendo apoptosis y promoviendo proliferación (fig. 4).


Algunas de las vías de señalización mayormente representadas por genes con alteración en la metilación en LLA son p53, WNT, EPHR, MAPK, y PI3K-AKT4,20,59, las cuales están estrechamente relacionadas a procesos celulares como los ya mencionados (fig. 4). Una de las vías más profundamente estudiadas es la de TP53, el cual es un gen supresor de tumores involucrado en la activación de reparación de DNA dañado, inducción del punto de control G1/S y promoción de la apoptosis74. A pesar de que las mutaciones en este gen se presentan en el 50% de los tumores, en la LLA se encuentran en menos del 3%; sin embargo, en la LLA existe alteración de la vía p53 a nivel epigenético59. Se ha observado hipermetilación de, al menos, un gen involucrado en la vía p53 en 78% de los pacientes con LLA. También se ha detectado hipermetilación de genes involucrados en apoptosis dependiente de p53, como AIFM2, APAF1, DBC1, miRNA34B, miRNA34C, PMAIP1, POU4F2, PPP1R13B, TP73, NOXA, AMID y ASPP1. De igual forma, se ha encontrado hipermetilación en genes como CDKN1A, CDKN1C, CDKN2A, POU4F1, que participan en el control del ciclo celular dependiente de p53, e hipermetilación de LATS2 y DAPK1, que participan en la regulación de esta misma vía4,20,59,71,75.
La vía WNT/β-catenina ha sido ampliamente implicada en diversos tipos de cáncer76. La activación de esta vía regula, entre otros procesos, la proliferación y diferenciación celular4. En la LLA se ha reportado alteración en la metilación en genes asociados a la vía WNT, como WNT5A, RSPO1 y APC36,72,77. Adicionalmente, se ha reportado en la LLA-B en pacientes con recaída, metilación aberrante en los genes sFRP2, sFRP4, sFRP5, WIF1, DKK3, sFRP1, PTPRO, FZ10 y DKK2; también de los inhibidores de la β-catenina, las cadherinas (CDH1, CDH11, CD13), y genes de la familia SOX (SOX2,3,8,9,11,14,21)54.
Otras vías de señalización afectadas en la LLA por alteraciones en la metilación del DNA incluyen a los receptores efrina (EPH), que son receptores tirosina cinasa que activan y regulan diversos procesos biológicos. En la LLA, se ha sugerido que los genes EPH pueden actuar como supresores de tumor y se ha reportado hipermetilación en receptores y ligandos, incluyendo EPHA2, EPHA4, EPHA5, EPHA6, EPHA7, EPHA10, EPHB1, EPHB2, EPHB3, EPHB4, EFNA1, EFNA3, EFNA5, y EFNAB136. Adicionalmente, se ha reportado en la LLA alteración en la metilación de genes de la vía de las MAP cinasas (MAP2K1, BRAF, ERK2), así como de la vía PI3K-AKT54.
4Modificación de histonas en LLALas histonas son proteínas implicadas en la organización del DNA dentro del núcleo. Existen cinco tipos de histonas: H1/H5, H2A, H2B, H3 y H4. Las histonas H2A, H2B, H3 y H4 forman los nucleosomas que empacan al DNA, mientras que la histona H1 se encuentra en los espacios entre los nucleosomas; la histona H5 está presente en regiones específicas del DNA. El estado de la cromatina depende de modificaciones post-traduccionales en las histonas e influye en los estados de transcripción génica. Se conocen numerosas modificaciones covalentes en las histonas, como la acetilación, la metilación y la fosforilación, que conducen a la represión o activación génica (fig. 1)4,9. Las modificaciones predominantes y más estudiadas son la acetilación de lisina y la metilación de lisina/arginina9.
Las principales enzimas encargadas de regular la modificación de histonas son las metiltransferasas (HMTs), las desmetilasas (HDMs), las acetiltransferasas (HATs) y las desacetilasas (HDACs)78. La combinación en la actividad de estas enzimas confiere un “código de histonas”, que regula la topología de la cromatina y la accesibilidad de los promotores y, por lo tanto, regula la actividad transcripcional y otros procesos como la replicación y reparación del DNA4,79. Algunas de las modificaciones de histonas asociadas a la cromatina abierta, y por lo tanto a la activación transcripcional, son la acetilación de lisina (K) en la histona H3 (H3K4, H3K14, H3K9, H3K27) y en la histona H4 (H4K5, H4K20) (fig. 1). Las enzimas encargadas de la acetilación de histonas son las HATs. La metilación de lisinas catalizada por HMTs puede activar o reprimir la transcripción; por ejemplo, la monometilación de H3K4, H3K79 y H3K36 activa la transcripción. Algunas marcas de cromatina cerrada y represión transcripcional son la trimetilación de lisina en H3K9 y H3K27 (fig. 1)9. Junto con la metilación del DNA, las modificaciones de histonas se han implicado en la etiología y progresión de diversos tipos de cáncer80. Se han encontrado mutaciones en diferentes enzimas modificadoras de histonas que llevan a la pérdida o ganancia de su función. En particular, la acetilación remueve la carga positiva en las histonas, lo que disminuye la interacción de los grupos fosfato del DNA (carga negativa) con la porción N-terminal de las histonas (carga positiva). Lo anterior conduce a un relajamiento en la cromatina, que se asocia con mayor accesibilidad al DNA, y por lo tanto a la actividad transcripcional aumentada81. Las enzimas HDACs incluyen a las HDAC1, a la HDAC11 y a las sirtuinas82. La reducción en la acetilación de histonas derivada de la sobreexpresión de enzimas HDACs es un evento común en distintos tumores, incluyendo leucemias81–84.
En la LLA pediátrica se ha identificado la sobreexpresión de enzimas desacetilasas de histonas, como HDAC1, HDAC2, HDAC3, HDAC6, HDAC7 y HDAC8, en comparación con células no leucémicas (fig. 2)84,85. Particularmente, se ha reportado la sobreexpresión de HDAC2 y HDAC5 en la LLA-B84,86, mientras que se ha detectado mayor expresión de HDAC1, HDAC4 y HDAC5 en la LLA-T84. En cuanto a pronóstico, la sobreexpresión de los genes HDAC1, HDAC2, HDAC3, HDAC4, HDAC7, HDAC9 y HDAC11 se ha asociado con un pronóstico desfavorable en LLA pediátrica84,85,87. Por otro lado, la acetilación en la histona H4 se ha propuesto como un marcador de pronóstico al diagnóstico y en la recaída. En adultos con LLA, se identificó que los niveles aumentados de acetilación en H4 correlacionan con un incremento en la supervivencia88. Adicionalmente, se ha reportado que ETV6-RUNX1 recluta HDACs en LLA-B, lo cual induce la remodelación de la cromatina, y en consecuencia bloquea la transcripción de ciertos genes normalmente activados por RUNX189.
A pesar de que aún no se han identificado todas las vías de señalización y procesos celulares en los que impacta la alteración de las HDAC mencionadas en LLA pediátrica, se conoce que están involucradas en procesos como la transcripción (HDAC1, HDAC3, HDAC4, HDAC6, HDAC7, HDAC8, HDAC9), regulación del ciclo celular (HDAC1, HDAC2, HDAC3, HDAC4, HDAC6, HDAC7), apoptosis (HDAC1, HDAC2, HDAC3, HDAC6, HDAC7) y vías de señalización de p53 (HDAC2) y Notch (HDAC1, HDAC3, HDAC7) (fig. 3)4.
En la LLA también se han identificado HATs con expresión alterada. Las principales HATs incluyen las familias GNAT, MYST y CBP/p30082. Algunas de estas proteínas se han visto involucradas en rearreglos cromosómicos capaces de conducir a la transformación leucémica a través de la alteración en la acetilación de histonas, y por lo tanto en la expresión génica90. Algunas de las HATs que se han identificado con sobreexpresión en LLA-B son KAT7, KAT2A, CREBBP (CPB o KAT3A) y KAT6B86. Esta modificación se ha asociado con procesos celulares como la regulación transcripcional, la proliferación y la apoptosis86. Adicionalmente, se ha observado que la KAT2A acetila y estabiliza a la oncoproteína TCF3-PBX1 en células LLA91. También se ha encontrado que las mutaciones del gen CREBBP, que conducen a una desregulación transcripcional de sus genes blanco, están asociadas con recaída en LLA-B pediátrica92,93. Se ha determinado que la sobreexpresión en CREBBP puede conferir mal pronóstico; incluso, en aquellos subtipos de LLA-B con riesgo estándar, como la hiperdiploidía93.
La HMT más estudiada en la LLA es MLL, la cual metila la lisina 4 de la histona H3 (H3K4), que es una marca de cromatina abierta, y por lo tanto favorece la transcripción4. Adicionalmente, MLL regula la transcripción a través del reclutamiento de proteínas HATs como CBP Y MOF. Como parte de los múltiples blancos de MLL, se encuentran principalmente los genes HOXA. El gen MLL forma rearreglos con más de 85 diferentes genes, que conducen a la formación de proteínas quiméricas; aunque pierden el dominio con actividad H3K4 metiltransferasa, retienen la capacidad de unirse a la cromatina94,95. Las oncoproteínas MLL conducen a la activación aberrante de sus genes blanco a través de diversos mecanismos. Por ejemplo, la proteína quimérica MLL-AF4 altera la regulación de sus genes blanco al reclutar complejos de regulación epigenética como SEC, y DOT1 (H3K79 metiltransferasa). Esta desregulación lleva a la transformación leucémica, ya que entre los blancos de MLL-AF4 se encuentran los genes MEIS1, RUNX1, FLT3, MYC, BCL2, y PROM196. Adicionalmente, MLL se ha encontrado fusionado con proteínas HATs, como CREBBP y EP300, llevando a la sobreexpresión de sus genes blanco96,97.
5Alteraciones en miRNASLos miRNAs constituyen otro tipo de regulación epigenética. Son pequeños RNAs de 18-25 nucleótidos que, en mamíferos, inhiben principalmente la traducción de sus mRNAs blanco a través de la interacción con la región 3’UTR. Los miRNAS derivan del procesamiento de precursores por medio del complejo proteico RNAsaIII-Drosha y Dicer98. La regulación de la expresión génica por medio de miRNAs es un proceso biológico común; más del 60% de los mRNAs pueden ser regulados por miRNAs99. Los miRNAs están implicados en procesos biológicos críticos, incluyendo el crecimiento y desarrollo celular, metabolismo, proliferación, diferenciación y apoptosis17. En cáncer, se ha observado la participación de diversos miRNAs actuando como oncogenes (oncomiRNAs), supresores de tumores y supresores o activadores de metástasis17. Adicionalmente, los miRNAs pueden ser regulados por metilación del DNA y por modificación de histonas (fig. 2)100.
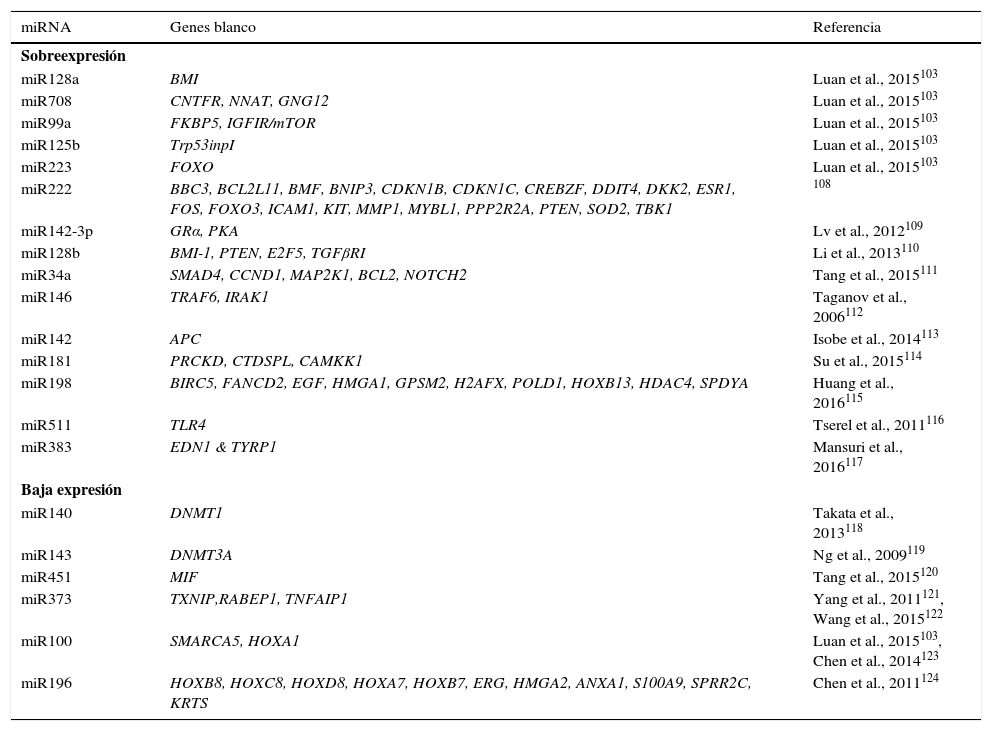
Múltiples procesos biológicos son regulados por miRNAs, incluyendo la hematopoyesis normal y maligna101,102. En LLA-B y T de niños y adultos, se han identificado alteraciones en la expresión de miRNAs17. Se han obtenido perfiles de expresión de miRNAs que se han utilizado en el diagnóstico, clasificación y pronóstico de la LLA103. En la LLA pediátrica, se ha reportado la sobreexpresión de miR222, miR339, miR142-3p, miR128a, miR128b, miR34a, miR146, miR142, miR181; así como la expresión disminuida de miR140, miR143, miR451, miR373, miR100, miR196b, en comparación con su contraparte celular normal (tabla 6). También se han identificado firmas de miRNAs que son distintivas entre los diferentes subtipos genéticos de LLA. En el subtipo hiperdiploide, se ha detectado la sobreexpresión de miR198, miR222, miR223, miRNA511 y miRNA708, mientras que el subtipo ETV6-RUNX1 presenta sobreexpresión de miR99a, miR100, miR125b, miR383 y miR708 (tabla 6)103–124. El subtipo de LLA con rearreglos en MLL se caracteriza por presentar sobreexpresión de miR196b y baja expresión de miR128b y miR221103,104.
Genes blanco de miRNAs frecuentemente alterados en LLA
| miRNA | Genes blanco | Referencia |
|---|---|---|
| Sobreexpresión | ||
| miR128a | BMI | Luan et al., 2015103 |
| miR708 | CNTFR, NNAT, GNG12 | Luan et al., 2015103 |
| miR99a | FKBP5, IGFIR/mTOR | Luan et al., 2015103 |
| miR125b | Trp53inpI | Luan et al., 2015103 |
| miR223 | FOXO | Luan et al., 2015103 |
| miR222 | BBC3, BCL2L11, BMF, BNIP3, CDKN1B, CDKN1C, CREBZF, DDIT4, DKK2, ESR1, FOS, FOXO3, ICAM1, KIT, MMP1, MYBL1, PPP2R2A, PTEN, SOD2, TBK1 | 108 |
| miR142-3p | GRα, PKA | Lv et al., 2012109 |
| miR128b | BMI-1, PTEN, E2F5, TGFβRI | Li et al., 2013110 |
| miR34a | SMAD4, CCND1, MAP2K1, BCL2, NOTCH2 | Tang et al., 2015111 |
| miR146 | TRAF6, IRAK1 | Taganov et al., 2006112 |
| miR142 | APC | Isobe et al., 2014113 |
| miR181 | PRCKD, CTDSPL, CAMKK1 | Su et al., 2015114 |
| miR198 | BIRC5, FANCD2, EGF, HMGA1, GPSM2, H2AFX, POLD1, HOXB13, HDAC4, SPDYA | Huang et al., 2016115 |
| miR511 | TLR4 | Tserel et al., 2011116 |
| miR383 | EDN1 & TYRP1 | Mansuri et al., 2016117 |
| Baja expresión | ||
| miR140 | DNMT1 | Takata et al., 2013118 |
| miR143 | DNMT3A | Ng et al., 2009119 |
| miR451 | MIF | Tang et al., 2015120 |
| miR373 | TXNIP,RABEP1, TNFAIP1 | Yang et al., 2011121, Wang et al., 2015122 |
| miR100 | SMARCA5, HOXA1 | Luan et al., 2015103, Chen et al., 2014123 |
| miR196 | HOXB8, HOXC8, HOXD8, HOXA7, HOXB7, ERG, HMGA2, ANXA1, S100A9, SPRR2C, KRTS | Chen et al., 2011124 |
Los diversos miRNAs implicados en la proliferación celular y apoptosis han sido asociados con el pronóstico de pacientes con LLA. La baja expresión de miR456, miR708, miR210, así como la sobreexpresión de miR100/99 han sido asociadas con resistencia a quimioterapia, y por lo tanto con un pronóstico desfavorable. Otros miRNAs cuya baja expresión se relaciona con mal pronóstico son miR124a y miR152. Adicionalmente, la alta expresión de miR92a está asociada con un pronóstico desfavorable60,65,125–127.
6Terapia epigenética en LLALa identificación de alteraciones epigenéticas en LLA ha motivado la búsqueda de moléculas capaces de revertir dichas alteraciones. En pacientes con LLA se han probado dos agentes hipometilantes, que fueron aprobados previamente en los Estados Unidos para su uso en otros tipos de cáncer. En pacientes con LLA refractaria o en recaída se han utilizado los inhibidores de la DNA-metiltransferasa, 5-azacitidina y decitabina, en combinación con fármacos quimioterapéuticos convencionales. En estos estudios se observó evidencia de actividad clínica sin toxicidad excesiva128,129. Además de los agentes desmetilantes, se han probado agentes inhibidores de histonas en pacientes con LLA, como el vorinostat y el panobinostat. En un estudio de fase II se probó la administración de decitabina en combinación con vorinostat antes de la quimioterapia de reinducción en niños y adultos con LLA refractaria. En 1/13 pacientes se registró muerte atribuida a toxicidad. En 5/8 pacientes que completaron el tratamiento, se realizó un trasplante alogénico de células hematopoyéticas; de estos, tres murieron por causas relacionadas con el trasplante y dos sobrevivieron sin evidencia de la enfermedad130.
Adicionalmente, se ha planteado que a través de la regulación de ciertos miRNAs se podría tener una mejora en el efecto terapéutico de los glucocorticoides, en particular para aquellos pacientes que muestran resistencia a agentes como la prednisona. Se ha demostrado que la inhibición del miRNA-17 lleva a un incremento en la sensibilidad de las células leucémicas a la dexametasona131. En aquellos pacientes con rearreglos en MLL insensibles al tratamiento con glucocorticoides, se ha sugerido la restauración de la expresión de miR128b y miR221 como terapia. Ambos miRNAS muestran baja expresión en pacientes con LLA con rearreglo en MLL. Se ha observado que miR128b tiene como blancos a los genes MLL, AF4, e incluso a las fusiones oncogénicas MLL-AF4 y AF4-MLL. Asimismo, miR222 es capaz de regular negativamente a CDKN1B. De manera cooperativa, ambos miRNAs son capaces de potenciar la sensibilidad de las células leucémias al tratamiento con glucocorticoides132. La restauración de la expresión del miR143 también ha sido propuesta como terapia para los pacientes con LLA con rearreglo en MLL; en estos pacientes, el miR143 se encuentra hipermetilado y silenciado. Este miRNA se ha identificado como un regulador de la expresión de MLL-AF4; la restauración de miRNA143 induce a apoptosis de las células leucémicas133.
En pacientes con LLA BCR-ABL1 positiva, se ha propuesto la restauración de la expresión del miR203 como terapia. Este miRNA tiene como blancos a ABL1 y a BCR-ABL1. En leucemias BCR-ABL1 positivas, se ha encontrado a miR203 silenciado por mecanismos genéticos y epigenéticos. En pacientes con LLA BCR-ABL1 positiva, que muestran resistencia a inhibidores tirosina cinasa, también se ha sugerido que podría utilizarse la restauración de miR217 como terapia para inhibir a la DNTM3A134,135. Se ha reportado que los pacientes BCR-ALB1 positivos adquieren resistencia a inhibidores de tirosina cinasa a través de la regulación a la baja de miR217 que, a su vez, regula a la alta a la DNMT3A.
7ConclusionesLa regulación epigenética a través de la metilación del DNA, de modificaciones en las histonas y de la regulación por medio de miRNAs juega un papel fundamental en el desarrollo y evolución de la LLA y otros tipos de cáncer. En la LLA, estos niveles de regulación han sido implicados en la etiología de la enfermedad, así como en el diagnóstico, clasificación y pronóstico de los pacientes. Con el advenimiento de los estudios amplios del genoma ha sido posible la detección de un número extenso de modificaciones epigenéticas características de la LLA, que reflejan la complejidad y heterogeneidad de la regulación epigenética en esta enfermedad. La investigación en este campo seguirá ofreciendo información que permita comprender mejor la compleja etiología de la LLA. Además, el conocimiento en la epigenética de la LLA contribuirá a identificar blancos terapéuticos que podrán ser modificados con tratamientos específicos, con la ventaja de que, a diferencia de las modificaciones genéticas, las alteraciones epigenéticas son reversibles.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
Fundación Miguel Alemán 2012, Fondos Federales 2013 Instituto Nacional de Pediatría, Fondo Sectorial de Investigación para la Educación SEP-CONACyT CB-2012-01/183467. Navarrete-Meneses agradece al Posgrado en Ciencias Biológicas, CONACyT 385279.