


Recién nacido masculino referido por enfermedad adenomatoidea quística.
1.1Antecedentes heredofamiliaresMadre de 21 años, estudiante de licenciatura, GI, CI, P0, A0, una pareja sexual. Padre de 21 años con preparatoria, empleado, consume cinco cigarros/día.
1.2Antecedentes personales no patológicosHabitan casa prestada con servicios básicos de urbanización para cinco personas en tres cuartos. Conviven con un perro. En ayuno desde el nacimiento. Ninguna inmunización.
1.3Antecedentes perinatales y patológicosProducto de GI con control prenatal desde el segundo mes y administración de ácido fólico, hierro y vitaminas. Infección de vías urinarias en el primer trimestre y cervicovaginitis en el tercero, sin especificar tratamiento. USG obstétrico a la semana 20 que identificó enfermedad adenomatoidea quística. Se realizó cirugía fetal con broncoscopia fetal diagnóstica. Se descartó atresia bronquial y se colocó derivación toraco-amniótica para drenar quistes. Por falla cardiaca fetal se realizó ablación láser de circulación sanguínea del tumor para disminuir su tamaño. Se administraron a la madre tres dosis de cefotaxima como profilaxis.
1.4Padecimiento actualPaciente que ingresó a este instituto con el diagnóstico fetal de enfermedad adenomatoidea quística. Nació por cesárea a las 40 SDG. Se intubó. Por frecuencia cardiaca<60 se dio un ciclo de compresiones torácicas, con lo que recuperó la frecuencia cardiaca. Apgar 3/7. Se colocaron accesos arterial y venoso umbilicales.
Exploración física.Recién nacido (RN) masculino con adecuada coloración.
Cabeza y cuello. Normocéfalo, fontanela anterior de 2×2cm, y posterior puntiforme. Pupilas isocóricas, pabellones auriculares bien implantados, narinas permeables. Se le colocó tubo endotraqueal de 3.5mm; el cuello cilíndrico sin adenomegalias. Ventilación mecánica convencional.
Tórax. Tórax simétrico, precordio con ruidos cardiacos rítmicos sin agregados. Campos pulmonares con hipoventilación generalizada, fístula paraescapular izquierda de 2cm.
Abdomen. Blando, depresible, sin visceromegalias. Genitales masculinos, sin alteraciones.
Extremidades. Íntegras, pulsos simétricos.
USG transfontanelar y renal. Sin evidencia de lesiones focales ni datos de sangrado; plexos coroideos bien definidos y simétricos. Núcleos de la base y fosa posterior normales. Riñones normales.
Ecocardiograma. Levocardia, retornos venosos normales, conexión atrio ventricular y ventrículo auricular concordante, presión de la pulmonar de 95mmHg. Insuficiencia tricuspídea grave, dilatación de las cavidades izquierdas, compresión externa del ventrículo izquierdo. Adecuada función ventricular. No se visualizó cortocircuito a través de foramen oval, y el conducto arterioso permeable.
Tratamiento. Lobectomía pulmonar superior izquierda. Se manejó con dopamina, adrenalina y norepinefrina. Ventilación con alta frecuencia y óxido nítrico inhalado a 20ppm. Tratamiento antimicrobiano con cefepime y amikacina. Se realizó toracocentesis derecha.
A pesar del manejo, presentó acidosis y paro cardiorrespiratorio sin respuesta a maniobras de reanimación.
2Presentación del caso2.1Dirección médica (Dr. Jaime Nieto-Zermeño)Se presenta un caso sui géneris que destacó por el apoyo por parte del Servicio de Cirugía Fetal y que constituyó todo un reto.
2.2Medicina fetal (Dr. Rogelio Cruz Martínez)Paciente de 21 años que a las 25 SDG se presentó con un feto único, varón, que venía referido de su médico ginecólogo por haber detectado un hidrops fetal y una masa pulmonar de lado izquierdo. En el ultrasonido obstétrico se observó una masa hiperecogénica con un gran componente sólido e imágenes quísticas>1cm de diámetro, ocupando prácticamente todo el tórax de lado izquierdo y desviando el mediastino y el corazón hacia el lado derecho del feto. Los pulmones se observaban totalmente colapsados, con un área pulmonar que equivalía al 15% con respecto a lo esperado para el peso gestacional, además de un poco de líquido en los pulmones que equivalía a un derrame pleural. En el corte sagital se observó gran cantidad de líquido de ascitis con el diafragma íntegro, y a nivel del tórax una gran masa hiperecogénica con componente sólido y componente quístico, derrame pleural y edema subcutáneo. Por lo tanto, era un feto con falla cardíaca por una gran masa unilateral que desviaba el mediastino a la derecha, hipoplasia pulmonar grave e hidrops fetal.
Las posibilidades de muerte intrauterina asociada con el hidrops son del 100% y, si se hacía una cesárea, el riesgo de muerte neonatal era excesivamente alto debido a que era un feto de 720g, 25 SDG, con hipoplasia pulmonar severa y falla cardiaca. Se le ofrecieron a la madre las siguientes opciones: 1) no realizar ningún procedimiento, con la seguridad de que habría muerte intrauterina; 2) realizar una cesárea, con el riesgo de muerte neonatal superior al 90%; o 3) realizar una cirugía fetal, con el 50% de posibilidades de éxito. La familia aceptó la tercera opción, que tenía las mayores posibilidades de éxito. Se propuso primero realizar una broncoscopia diagnóstica para descartar que hubiera comunicación con el árbol traqueobronquial y confirmar que era una malformación adenomatoidea macroquística (MAQ). Después, en el mismo procedimiento, con cirugía de mínima invasión, anestesia local a la madre y una aguja que atraviesa el útero, las membranas y la parte central e interior de estos quistes, colocar un catéter de derivación dentro de los quistes y con el otro extremo en la cavidad amniótica. Esto con el objetivo de que estos quistes, que estaban comunicados entre sí en su interior, se pudieran vaciar y disminuyera el tamaño del tumor, y así la compresión cardiaca asociada. Técnicamente la intervención fue exitosa, no hubo ninguna complicación o ruptura. El corazón se vio menos comprimido, pero persitió la gran masa sólido-quística y una gran hipoplasia pulmonar. La falla cardiaca empezó a disminuir y mejoró; sin embargo, la hipoplasia pulmonar persistió debido a la masa tumoral. Esta MAQ tenía flujo sanguíneo que derivaba de la arteria pulmonar. Por eso se propuso una segunda intervención para eliminar esta circulación sanguínea y disminuir el componente sólido de la MAQ, con la finalidad de aumentar el tamaño del pulmón del lado derecho. Se hizo la intervención una semana después de la primera, a las 26 SDG; otra vez, una cirugía de mínima invasión, percutánea, con anestesia local y anestesia intramuscular fetal, con fentanil, vercuronio y atropina, para que el feto no se moviera y no le doliera. Se hizo una ablación láser de toda la circulación sanguínea de la masa. La intervención fue exitosa en el sentido de que no hubo ninguna complicación. La falla cardiaca revirtió completamente, y para las 30 SDG ya no tenía derrame pleural ni ascitis. El pulmón derecho empezó a mejorar, a aumentar su tamaño y la masa tumoral empezó a disminuir. A las 32 SDG comenzó a disminuir el componente sólido de la masa, pero a aumentar el componente quístico. Se cree que el catéter que todavía tenía se obstruyó, y al obstruirse aumentó la cantidad de líquido dentro de la masa tumoral, pero el componente sólido que había disminuido gracias al láser intratumoral hizo que mejorara el tamaño tumoral del lado derecho, y que ya no tuviera falla cardiaca. Por eso se pensó que era más riesgoso hacer una tercera intervención fetal. En el ultrasonido de las 39 SDG ya no se veía líquido de ascitis. A nivel del tórax ya tenía un pulmón de mejor tamaño; el componente quístico de la masa empezó a aumentar de tamaño, pero el tamaño pulmonar derecho equivalía ya al 90% con respecto a lo esperado para la edad gestacional. Finalmente, las posibilidades de vida para este bebé a las 39 SDG eran mucho mejores con respecto a las que tenía a las 25 SDG.
Esta es la situación en el manejo fetal, el niño nació, y a continuación se describen los hallazgos radiológicos.
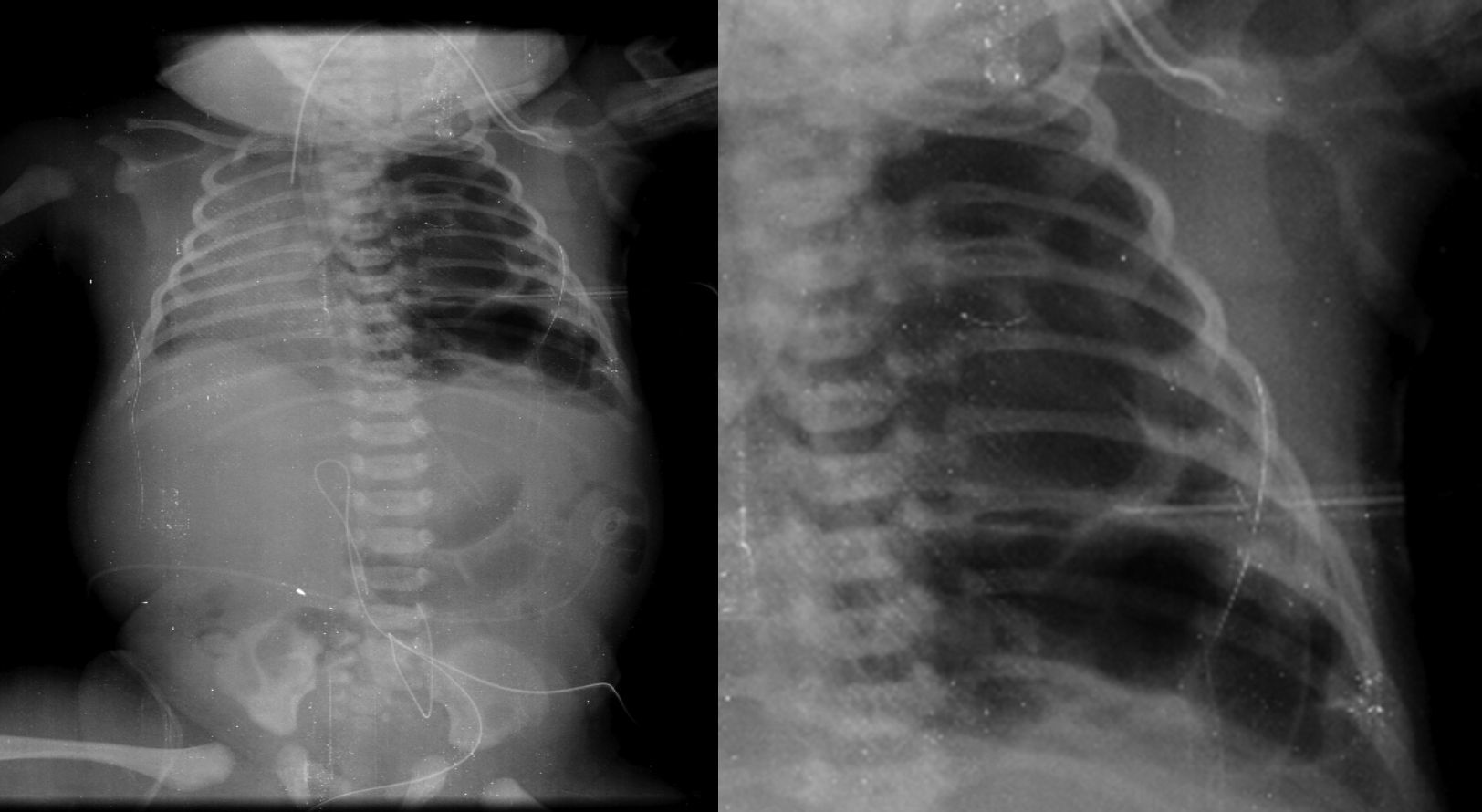
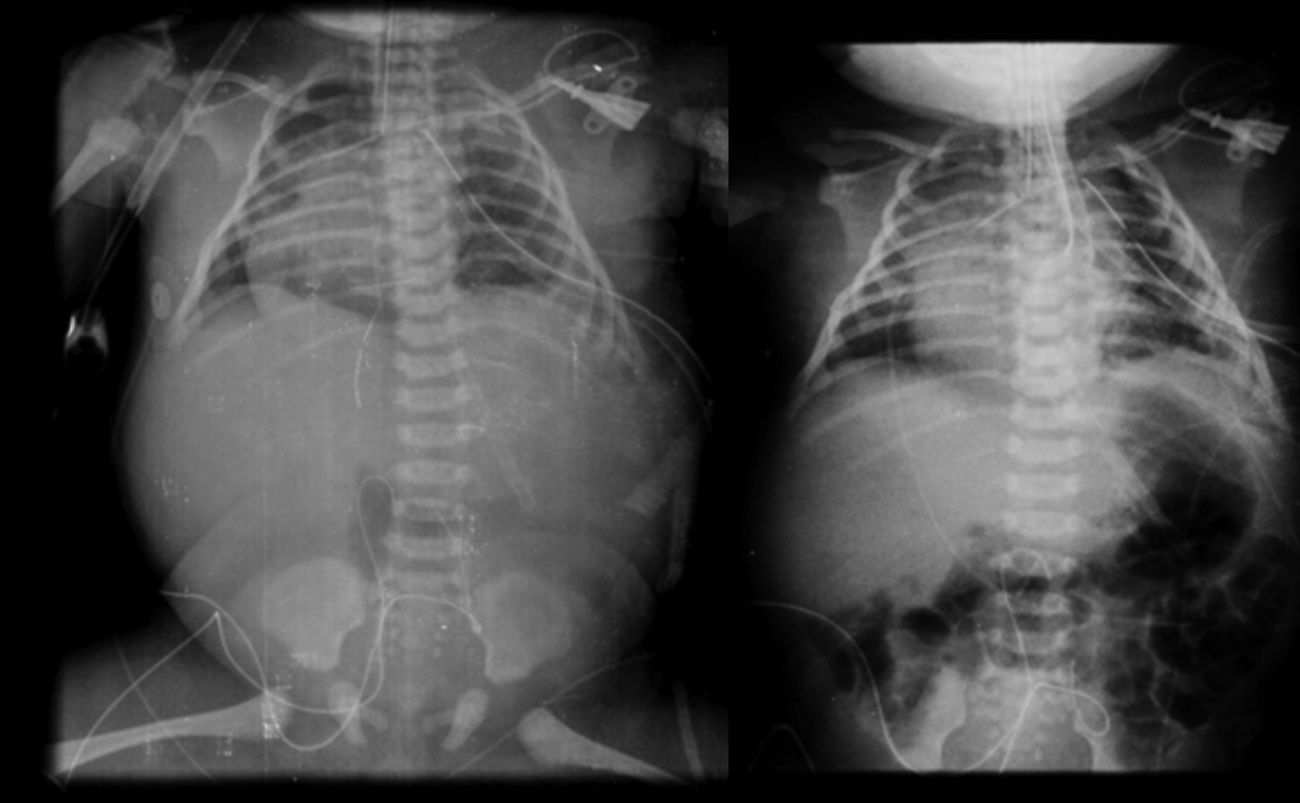
2.3Imagenología (Dra. Bertha Romero-Baizabal)La primera radiografía (24 de enero del 2013) es la más demostrativa. Se observa el pulmón izquierdo lleno de imágenes radiolúcidas, bien circunscritas, de gran tamaño y que ocupan prácticamente todo el pulmón izquierdo. Las estructuras del mediastino así como el mismo pulmón izquierdo están desplazados hacia el lado derecho (Figura 1). El diafragma se observa aparentemente íntegro y hay poca distribución de gas intestinal. La cánula traqueal se encuentra en buena situación, pero lateralizada hacia la derecha. En una secuencia (3 de febrero) se observó una imagen radiolúcida a nivel basal de lado izquierdo en relación con un neumotórax que persistió, aunque de menores dimensiones. En el pulmón derecho se observaron cambios en la densidad en relación con un patrón de tipo reticular, lo cual no descartó la posibilidad de un proceso infeccioso. Por lo tanto, los diagnósticos radiológicos fueron una malformación congénita pulmonar de la vía aérea tipo I, un proceso neumónico pulmonar bilateral y neumotórax izquierdo. Las últimas placas fueron tomadas después de la resección del lóbulo superior del pulmón. Quedó un pulmón izquierdo muy pequeño, pero permitió que el pulmón derecho se reexpandiera. Quedó un neumotórax residual que es común en estos pacientes (Figura 2).


Se discute el caso de un paciente con MAQ, cuyos antecedentes de falla cardiaca fetal e hidrops nos colocan ante un paciente con un manejo y un tratamiento novedoso a una malformación habitual. Este paciente presentó los siguientes diagnósticos sindromáticos:
- •
Síndrome de dificultad respiratoria. Por aumento de la frecuencia respiratoria, hipoventilación generalizada, hipoxemia, hipercapnia y acidosis respiratoria.
- •
Síndrome de fuga aérea. Caracterizado por hipoventilación, timpanismo y la imagen radiográfica que evidencia un neumotórax.
- •
Síndrome de respuesta inflamatoria sistémica. Caracterizado por taquicardia persistente, fiebre de 38°C y leucopenia.
- •
Síndrome de condensación. Porque cursó con estertores finos en hemitórax derecho, matidez a la percusión y ausencia de ruidos en la base izquierda.
- •
Insuficiencia renal aguda. Por aumento de la creatinina de hasta 3.3mg/dl.
- •
Acidosis metabólica persistente.
Con base en lo anterior, los diagnósticos nosológicos son los que se mencionan a continuación:
- 1)
Malformación adenomatoidea quística, cuyo crecimiento condicionó la compresión cardiaca fetal con un hidrops no inmune, la falla cardiaca y el neumotórax recidivante.
- 2)
Hipertensión arterial pulmonar, con presión pulmonar cuantificada hasta en 95mmHg, la cual causó hipoxemia persistente, y no respondió a la administración de aminas inotrópicas, vasodilatadores pulmonares y óxido nítrico.
- 3)
Infección asociada al cuidado de la salud, con neumonía asociada con el ventilador y con la manipulación intrauterina de la vía aérea, y también a la intubación orotraqueal desde el nacimiento, complicada con choque séptico por los datos de respuesta inflamatoria sistémica, que requirió apoyo aminérgico sin respuesta adecuada.
- 4)
Falla orgánica múltiple, por insuficiencia respiratoria, cardiovascular, renal y hematológica.
Es importante recalcar tres puntos principales del caso: el manejo prenatal, el manejo médico y el tratamiento quirúrgico. En relación con el manejo prenatal, se realizó en la semana 25 de gestación. Esto es muy importante, ya que el diagnóstico prenatal de este tipo de malformaciones solamente se realiza en el 20-30% de los embarazos, lo que permitió llevar a cabo el seguimiento, identificar los factores de mal pronóstico, como el hidrops fetal y la falla cardiaca en un feto menor de 30 SDG, y tomar la decisión de llevar a cabo la intervención fetal. El equipo perinatal moderno incluye a los médicos maternofetales y a los expertos en el diagnóstico de imagen, por ultrasonido o por resonancia magnética1,2. Además, es importante realizar un ecocardiograma por un subespecialista en ecocardiografía fetal. Los procedimientos quirúrgicos los puede realizar un médico maternofetal o un cirujano pediatra con entrenamiento en cirugía fetal. Estos casos se deben discutir junto con los neonatólogos que atenderán el nacimiento y los posibles subespecialistas involucrados en el manejo, para planear el sitio del nacimiento, los procedimientos que se llevarán a cabo durante la reanimación y la unidad de cuidados intensivos neonatales, y el tiempo ideal para realizar el procedimiento quirúrgico definitivo. En este caso, primero se llevó a cabo la derivación toracoamniótica. La serie de casos más grande reportada con este tratamiento para MAQ es de tan solo 11 pacientes. El procedimiento ha demostrado incrementar la sobrevida de los pacientes del 33 al 75%. En todas las series reportadas, el factor de peor pronóstico ha sido la presencia de hidrops fetalis3. Después de la derivación, el principal parámetro para evaluar la mejoría del feto es la medición de la relación pulmón-perímetro cefálico. Posteriormente se llevó a cabo la ablación con láser porque el feto cursó con falla cardiaca. Se revisó la literatura y se encontraron solo 16 pacientes con malformación pulmonar que se trataron con ablación por láser, de los cuales 9 cursaron con secuestro pulmonar y solo 7 de ellos con una MAQ, reportando una sobrevida del 28.6%. Actualmente no existen guías de terapia prenatal para este tipo de lesiones fetales. Se han propuesto diferentes alternativas como la cirugía fetal abierta, la administración de esteroides por punción intrauterina, la embolización con alcohol, la simple aspiración de los quistes o incluso la administración de OK432. En el Hospital Infantil de México Federico Gómez se han atendido a los primeros neonatos con antecedente de intervención fetal en México, y este último es el único caso con MAQ.
En relación con el tratamiento médico, el paciente nació de término, presentando datos dificultad respiratoria desde su nacimiento, por lo que ameritó intubación orotraqueal y parámetros altos de ventilador. Cursó con una presión pulmonar de 95mmHg, con hipoxemia persistente, por lo cual se considera que, aunado a la ventilación de alta frecuencia oscilatoria, era necesario el uso de oxido nítrico y sildenafil desde un principio, así como otras alternativas terapéuticas, como la vasopresina en dosis bajas. Por el momento no se cuenta con oxigenación por membrana extracorpórea en la institución, lo cual hubiera sido una herramienta útil. Además, como ya se mencionó, se presentó un paciente potencialmente infectado por la manipulación intrauterina y con necesidad de cuidados intensivos. El manejo aminérgico fue muy dinámico porque el reto era mantener un presión arterial sistémica adecuada y disminuir la presión pulmonar. Este manejo fue guiado por monitoreo continuo mediante oximetría de pulso, gasometrías arteriales y venosas, y ultrasonido cardiaco adicional4–6.
Se debe recordar que la primera finalidad en el periodo neonatal inmediato es el drenaje de colecciones líquidas o de aire y ventilación de apoyo con corrección del estado hemodinámico. Se consideró adecuada la colocación de la sonda pleural y las maniobras necesarias para la correcta funcionalidad, ya que el paciente cursó con mala respuesta al tratamiento conservador. Se llevó a cabo la resección de la lesión, esperando una mejoría clínica por la disminución de la compresión del ventrículo izquierdo, con disminución transitoria de la presión pulmonar de hasta 30mmHg.
Por lo anteriormente discutido, los diagnósticos clínicos finales fueron los siguientes: recién nacido de término con peso adecuado para la edad gestacional, antecedentes de hidrops no inmune y falla cardiaca fetal, malformación adenomatoidea quística de tipo 1, hipertensión pulmonar severa, neumonía nosocomial, choque séptico refractario a aminas y falla orgánica múltiple.
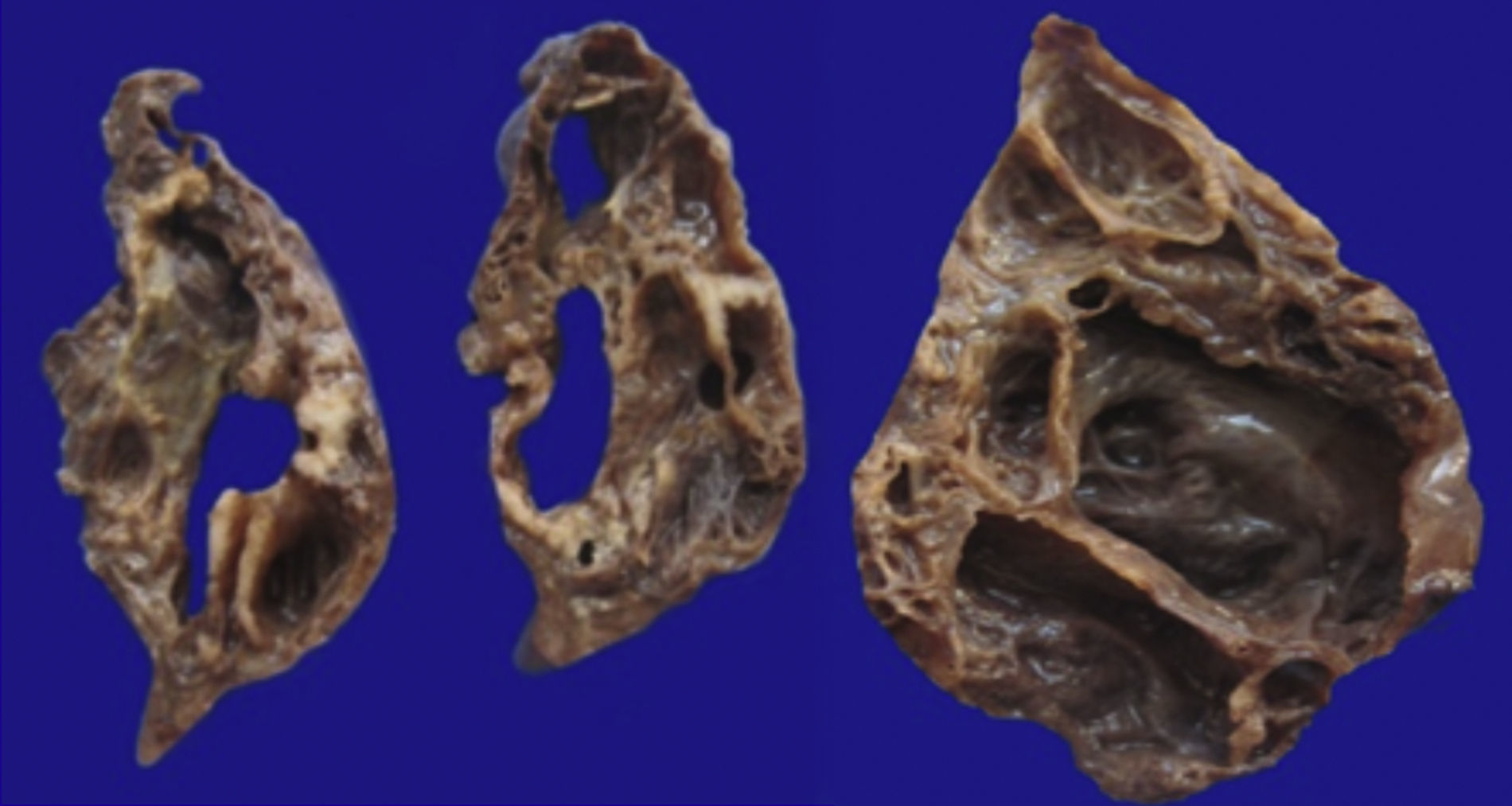
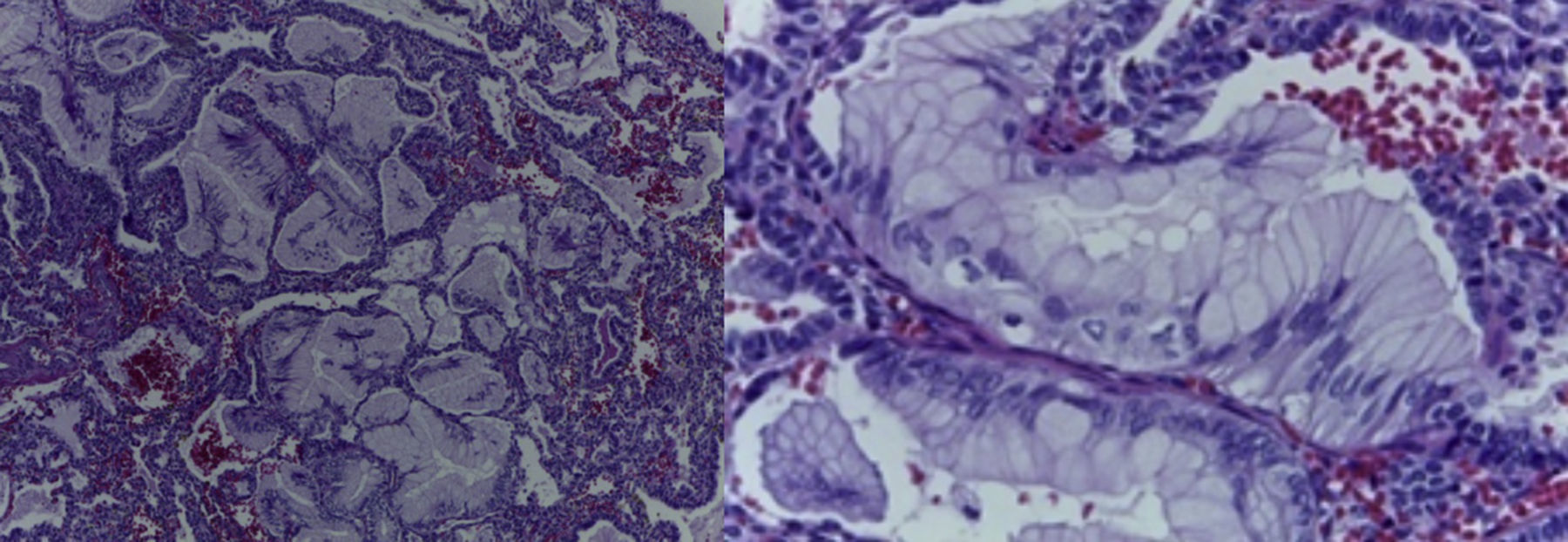
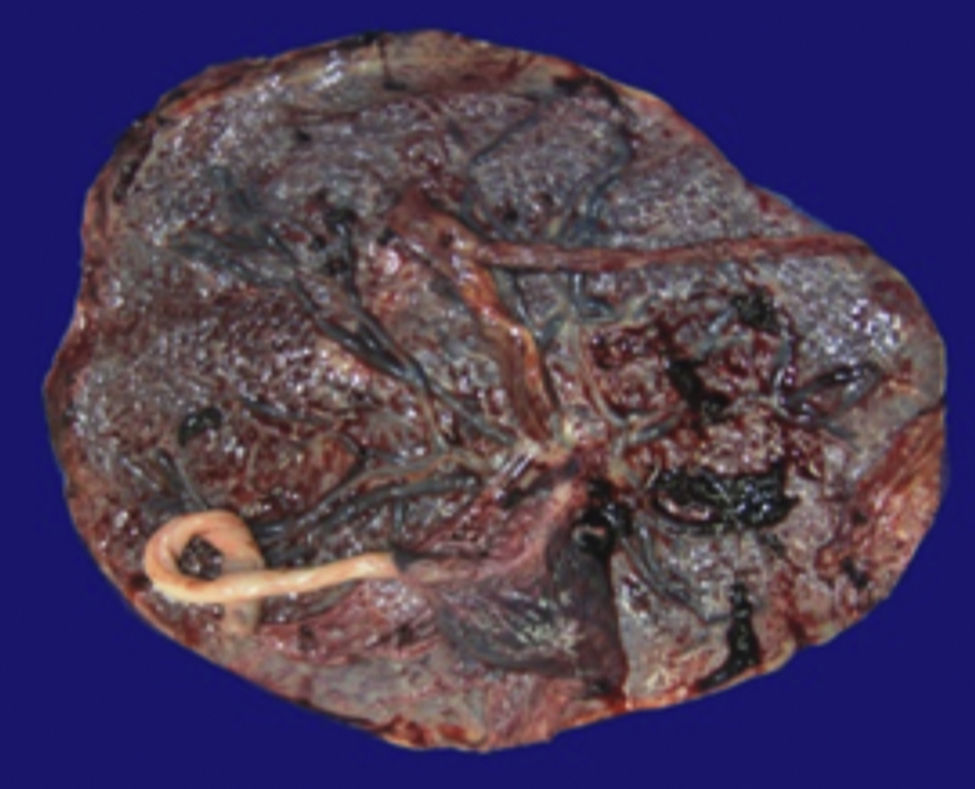
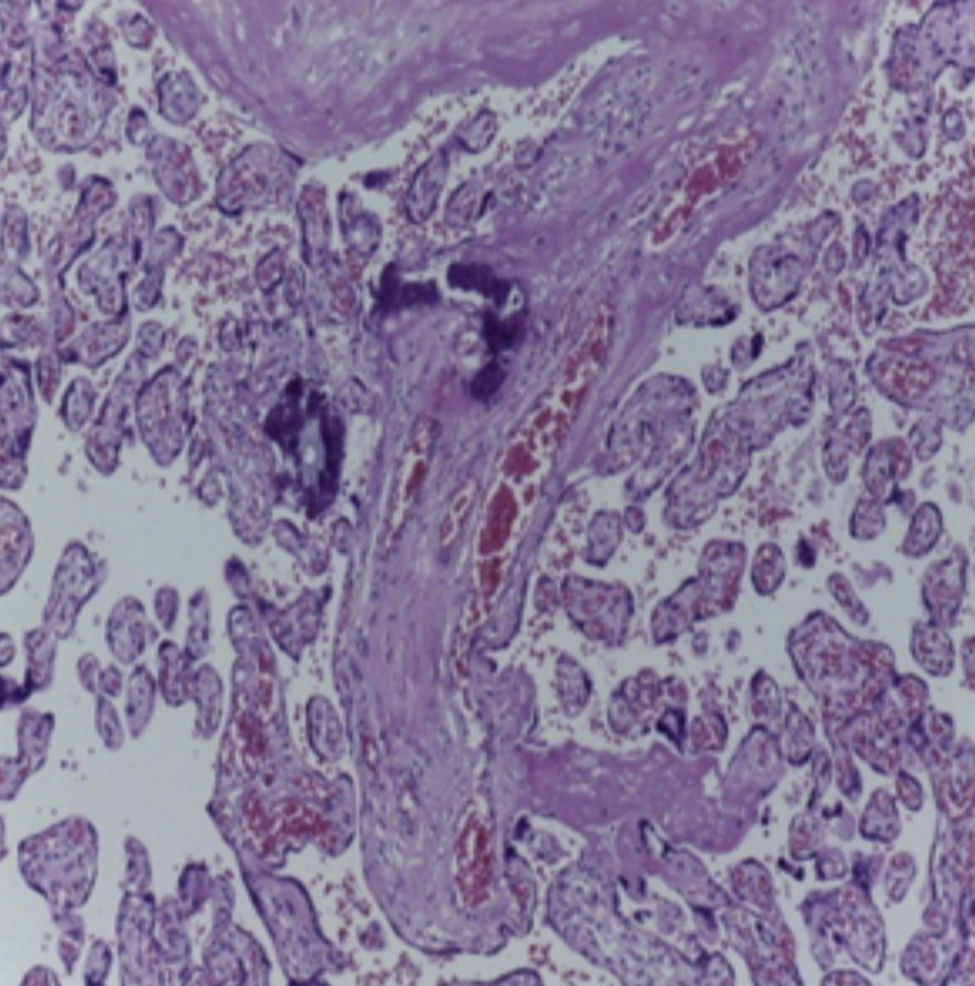
4Patología (Dra. María Argelia Escobar Sánchez)Se observa una pieza quirúrgica que corresponde al lóbulo superior del pulmón izquierdo. Pesó 80g y presentaba una pleura opaca, despulida y congestiva. A los cortes seriados, se observaron numerosos quistes con un diámetro promedio entre 2 y 5cm. Entre los quistes había áreas de parénquima pulmonar con congestión importante. Existe un quiste de mayor tamaño, con diámetro de 6cm, cuya superficie interna es brillante y trabeculada (Figura 3). Otros quistes más pequeños estaban separados por tabiques de tejido conectivo, lo que se corroboró mediante la histología. La malformación estaba constituida por numerosos quistes revestidos por epitelio columnar y epitelio cilíndrico ciliado, que alternaban con algunas células mucosas. Estas células mucosas recuerdan las glándulas intestinales, mucoproductoras; entre estas se observaron tabiques de tejido fibroconectivo con algunas células comprimidas. Entre los quistes revestidos por este epitelio vemos, además, la presencia de músculo liso (Figura 4).
.")
.")
Además del producto de la lobectomía superior izquierda, también se recibió la placenta, que pesó 700g y midió 21×20cm. Las membranas estaban íntegras. Por la cara materna se pudieron observar los cotiledones sin alteración y por la cara fetal las membranas congestivas con inserción central del cordón. Era una placenta monoamniótica, monocoriónica (Figura 5). Los diferentes cortes de la placenta mostraron el cordón con vasos sanguíneos sin alteraciones, las membranas íntegras y congestivas, y la placenta con vellosidades del tercer trimestre del embarazo, con presencia de algunos trombos hialinizados y calcificaciones propias de la edad gestacional (Figura 6).


Con estos hallazgos, se realizó el diagnóstico de malformación pulmonar congénita de la vía aérea tipo I, o lo que antes se llamaba enfermedad adenomatoidea quística, y placenta del tercer trimestre del embarazo con calcificaciones, sin alteraciones. El habitus exterior mostró un producto del sexo masculino con peso de 3,500g contra un esperado de 3,100g. Presentó extensas áreas de descamación, una herida suturada a nivel de tórax, además de dos orificios que correspondían a sondas pleurales localizadas en el quinto y en el décimo espacio intercostal izquierdo. Al momento de la evisceración, ya no se encontró la presencia de neumotórax.
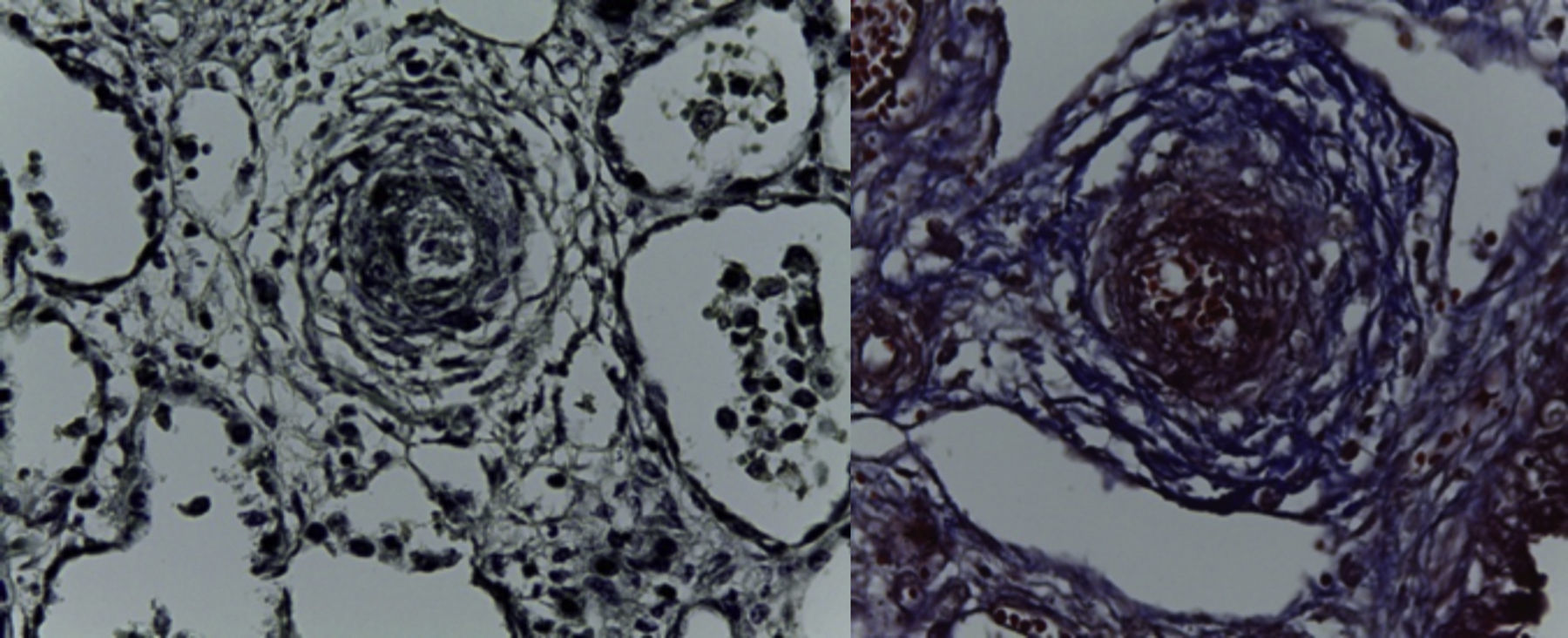
En la pieza que correspondía al bloque cardiopulmonar, se pudo observar el corazón con los grandes vasos de características normales, la pulmonar anterior, la aorta posterior, la circulación venosa pulmonar normal. Llama la atención una dilatación importante del ventrículo derecho. El pulmón derecho mostró sus tres lóbulos sin alteraciones, y se observó únicamente el pulmón residual izquierdo posterior al evento quirúrgico. A mayor aumento se pudo observar que la superficie pleural era anfractuosa, congestiva, y los hilos de sutura normales, sin presencia de hemorragia; es decir, no había alteraciones posteriores a la cirugía. En el corte correspondiente al diafragma se pudo observar congestión importante, e infiltrado inflamatorio constituido por linfocitos y células gigantes multinucleadas de tipo reacción a cuerpo extraño. La pleura también mostraba engrosamiento, infiltrado inflamatorio linfocitario y numerosas células gigantes multinucleadas que se extendían al parénquima pulmonar residual, donde se observaron los alveolos están colapsados y la presencia de granulomas secundarios al evento quirúrgico. El pulmón derecho mostraba infiltrado inflamatorio intraalveolar constituido por neutrófilos y algunas áreas de necrosis. Es decir, este paciente también presentaba un proceso infeccioso en el pulmón derecho. Había colapso de algunos alveolos, y en otras áreas había hemorragia pulmonar extensa además de edema intraalveolar y presencia de hipertensión pulmonar vascular localizada en las arterias y arteriolas de pequeño calibre menores a 200μm. En estas se pudo ver un aumento en el tejido conectivo en la adventicia, lo cual fue evidente con las tinciones especiales de Masson y fibras elásticas (Figura 7), como la hipertrofia del músculo e hiperplasia de las células del endotelio. El parénquima pulmonar residual presentaba hipoplasia; es decir, menos de seis alveolos, contando desde la pleura hasta el bronquiolo terminal. Estos presentaban compresión, por lo que había proliferación celular en el intersticio. El corazón tuvo un peso de 50g contra un esperado de 20g, y presentaba hipertrofia y dilatación importante de las cavidades derechas. A la apertura de las cavidades se encontró un foramen oval de 0.2cm de diámetro. Histológicamente, solo se observaron algunos núcleos hipertróficos de los cardiomiocitos, y el resto sin alteraciones. El hígado estaba aumentado de tamaño, con un peso de 200g contra un esperado de 127g; macroscópicamente, con áreas de congestión. Histológicamente se observó una congestión importante de los sinusoides localizados en el lobulillo y alrededor de las venas centrales. A mayor aumento se identificó extravasación de eritrocitos, es decir, una obstrucción al flujo venoso.
.")
Con estos hallazgos, se diagnosticó una malformación congénita de la vía aérea tipo I por el tamaño de los quistes y por la presencia de músculo liso localizado entre los quistes. Cabe mencionar que esta malformación es la que se presenta con mayor frecuencia, aproximadamente en el 70% de los casos. Anteriormente se describían tres tipos. Ahora se sabe que son cuatro. Stocker los describió a partir del 2002, y se clasifican del 0 al 4. El cero es incompatible con la vida, y el cuatro es el extremo.
El paciente presentaba un estado postlobectomía superior izquierda, la pleuritis aguda y crónica con reacción granulomatosa a cuerpo extraño, una neumonía basal derecha y la enfermedad vascular pulmonar grado B de Rabinovitch. Además, cardiomegalia, el foramen oval permeable, había persistencia del conducto arterioso permeable de 0.3cm, insuficiencia cardiaca congestiva, hepatomegalia congestiva y la obstrucción al flujo venoso.
El sistema nervioso central tuvo un peso de 450g, contra un esperado de 385g. Macroscópicamente, los cortes de la porción supratentorial mostraban ligera dilatación del sistema ventricular, pero no había presencia de hemorragia de la matriz germinal ni otras alteraciones. Histológicamente, los cortes de la corteza mostraron un desarrollo normal, con una migración cortical normal para la edad, y había algunas neuronas de aspecto hipóxico con retracción importante del citoplasma e hipercromasia y picnosis de los núcleos.
Los riñones tuvieron un peso de 16g, contra un esperado de 20g. Por su superficie capsular presentaban lobulaciones, que es normal hasta los dos años de edad. Se contabilizaron siete cálices renales, lo que eliminó el diagnóstico de una hipoplasia renal. Lo que sí fue evidente era la hipotrofia por el peso. Hubo congestión importante de las papilas a nivel de la médula. Histológicamente se pudo observar la corteza de características normales. Ya no había glomerulogénesis (los glómerulos eran de término). Había congestión importante en los vasos localizados en la corteza glomerular. A mayor aumento se pudo confirmar lo anterior, además de la pérdida del epitelio de los túbulos corticales secundaria al estado de choque.
En el tubo digestivo el esófago no presentaba alteraciones; la mucosa gástrica con congestión. Sin embargo, histológicamente estaba íntegra y no había más que bandas de contracción a nivel de la musculatura lisa. El intestino delgado y el colon mostraron algunas áreas de congestión a nivel de la mucosa. Histológicamente había algunas zonas de congestión en la lámina propia y autolisis. En todos los cortes hubo presencia de neuronas ganglionares en los plexos submucosos y mientéricos. En los cortes de colon se pudo ver la congestión secundaria al choque, la contracción y la presencia de neuronas ganglionares. En el páncreas no hubo alteraciones histológicas; los acinos y los islotes de Langerhans sin alteraciones. El bazo tuvo un peso de 15g, contra un esperado de 12g. Macroscópicamente presentaba áreas de congestión, que se corroboraron desde el punto de vista histológico, y una disminución importante del tejido linfoide. La médula ósea de características normales, con osificación endocondral que corresponde a un niño de la edad de este paciente, y sin alteraciones. Los ganglios no tenían todavía los folículos secundarios; únicamente primarios. El timo se observó con algunos corpúsculos de Hassall dilatados y calcificados, con ligera disminución del tejido linfoide. La glándula suprarrenal estaba íntegra, con congestión a nivel de la médula pero sin hemorragia. En el testículo había congestión intersticial. Todos los cultivos post mórtem fueron negativos.
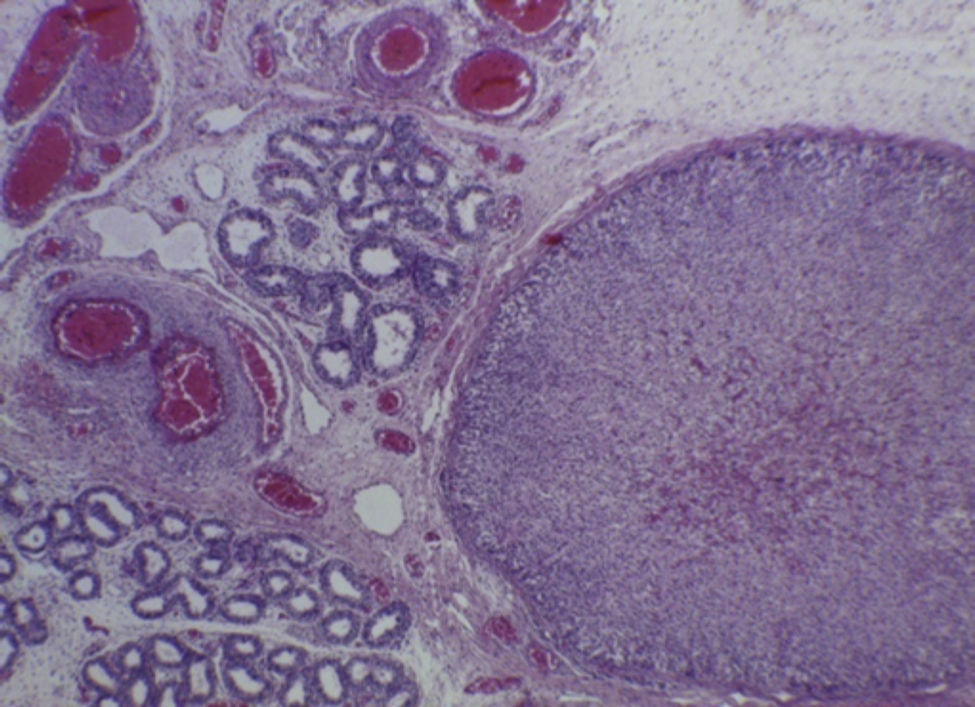
Con estos hallazgos se integraron los diagnósticos de edema cerebral, hipotrofia renal bilateral, y congestión multivisceral, que es uno de los datos anatómicos de choque. La hipertrofia ventricular derecha de larga evolución fue resultado de toda la alteración que presentó durante la vida fetal, y esto fue reflejo de la hipertensión pulmonar persistente in útero. Como hallazgo independiente se encontró una glándula suprarrenal localizada en el epidídimo del testículo derecho, que no tiene ninguna traducción clínica (Figura 8)7–10.
5Comentarios finales5.1Neurología (Dr. Saúl Garza Morales).")
La medicina fetal ha cambiado y la forma de abordaje también. Uno de los puntos que se discutía anteriormente fue con respecto a los equipos de trabajo que iban a hacerse cargo del RN, y los equipos de trabajo que trabajaban con el feto. Es decir, la importancia de formar equipos interdisciplinarios con perinatólogos, neonatólogos, cirujanos postnatales, trabajando en conjunto. Esto resulta de suma importancia para este y otro tipo de malformaciones, como por ejemplo las de vías urinarias. Cuando se atiende un paciente prenatal, otros estudios básicos son los de tipo cromosómico o genético.
5.2Dirección médica (Dr. Jaime Nieto-Zermeño)Se realizó el estudio genético, y además se contó con el trabajo entusiasta de neonatólogos y perinatólogos. Antes de la cirugía fetal, siempre se discute el caso con el Dr. Rogelio Cruz Martínez que radica en Querétaro. Se cuenta con el apoyo total e incondicional del grupo de Cirugía Fetal del Hospital General de México, del grupo de Anestesia y de Cirugía General Pediátrica.
Este paciente ilustra claramente la posibilidad de lograr algo, ya que el pronóstico inicial era de mortinato; es decir, no hubiera podido nacer. A pesar del esfuerzo, no se logró que el paciente sobreviviera.
5.3Neonatología (Dr. Daniel Ibarra Ríos)Es muy importante el manejo interdisciplinario y la comunicación, para poder llevar a cabo estos procedimientos. A partir de casos con el 100% de mortalidad in útero, se está viendo qué se puede hacer y qué procedimientos ofrecer.
Sin embargo, se necesita tener todo bajo control, y lo más difícil es la logística. Se comentan los casos con Cirugía Fetal, y a partir de ahí se realiza un gran trabajo con los servicios de enfermería, terapia respiratoria, insumos, anestesiología, etcétera.
Como se explicó anteriormente, al nacer, el niño se intuba con cordón abierto; tiene que haber tres o cuatro neonatólogos que van a recibirlo en la cuna de calor radiante. Existe una estrecha relación entre todos los servicios que van a acudir a la cuna del paciente que se encuentra en estado de gravedad. En estos niños, que al principio tienen hidrops, el control de la hipotensión es muy difícil. Primero, se deben estabilizar hemodinámicamente, atender la hipotensión y la hipertensión vascular pulmonar, y tomar muestras de líquido amniótico, además de enviar la placenta a Patología. Por otro lado, la parte infecciosa es muy complicada. No se cuenta con la experiencia necesaria sobre las infecciones relacionadas con la manipulación.
Lo más importante es estar al pendiente de la frecuencia cardiaca, y de la necesidad de compresiones torácicas. Deben estar otras dos personas disponibles para colocar los accesos vasculares. Se tiene la oportunidad del acceso a los vasos umbilicales, lo que permite la monitorización venosa y arterial. De ahí, se debe trasladar a la Unidad de Cuidados Intensivos Neonatales. Se manejan con ventilación convencional y de alta frecuencia. Se deben tener disponibles el óxido nítrico, las esterasas 3 y 5 y la vasopresina de rescate para el manejo de la enfermedad vascular pulmonar. Es también importante el reconocimiento temprano de la insuficiencia suprarrenal para, de ser necesario, iniciar el manejo con esteroides, la medición de cortisol, etcétera. En resumen, es un trabajo muy complicado. Se está ya en vías de formalizar listas de cotejo, y un trabajo bien estandarizado para ver qué opciones se pueden ofrecer. También es importante la intervención de trabajo social y psicología, sobre todo por el apoyo que se tiene que brindar a los padres. Este trabajo es importante, ya que incluso se logró obtener el estudio post mórtem, que ayuda para adquirir experiencia en estos casos.
Se han podido perfeccionar, sobre todo, los tiempos de intubación, el manejo de la hipertensión, etcétera, para afianzar el protocolo de hipertensión vascular pulmonar. Estos son los pacientes que antes no se veían, porque hay algunos casos donde la enfermedad adenomatoidea quística se revierte in útero. Por eso está aceptado que se apliquen esteroides. Lo importante es adquirir experiencia para que cambie de forma radical el manejo de estos niños. Ya se está en el camino gracias al advenimiento de la cirugía fetal.
5.4Cirugía Pediátrica (Dr. Ricardo Ordorica)La evolución, el diagnóstico y tratamiento temprano permiten conocer más casos. La medicina perinatal se plantea en qué casos se debe intervenir y cuáles deben llegar a término. Se considera al feto como un paciente que tiene derecho a la salud y a un tratamiento, en caso de que exista. En el hospital ya se puede ofrecer un tratamiento completo. Este caso en particular planteó retos para considerar cuál es el momento oportuno para operarlos.
5.5Dirección médica (Dr. Jaime Nieto-Zermeño)En cuanto a que si era factible un trasplante pulmonar, en realidad era imposible.
Este es un caso que deja una gran enseñanza.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que los procedimientos seguidos se conformaron a las normas éticas del comité de experimentación humana responsable y de acuerdo con la Asociación Médica Mundial y la Declaración de Helsinki.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.