





La enfermedad de Alexander consiste en una forma de leucodistrofia poco frecuente que afecta principalmente a los astrocitos; tiene un patrón de herencia autosómica recesiva y es causada por mutaciones en el gen GFAP, localizado en el cromosoma 17q21. Puede presentarse a cualquier edad y la forma infantil se caracteriza por macrocefalia, crisis convulsivas, retraso motor y cognitivo grave y espasticidad o ataxia progresivas.
Caso clínicoPaciente de sexo femenino de 8 meses evaluada por retraso psicomotor y crisis convulsivas motoras focales no provocadas. En la exploración física, con perímetro cefálico normal, respuesta motora incrementada ante estímulos táctiles y al ruido, signos piramidales y ausencia de visceromegalias. Se observó hipodensidad generalizada de la sustancia blanca en la resonancia magnética y punción lumbar con hiperproteinorraquia. Se descartó enfermedad de Krabbe mediante ensayo enzimático y secuenciación del gen GALC. En la reevaluación del caso, las alteraciones en la neuroimagen hicieron sospechar de enfermedad de Alexander, y la secuenciación del gen GFAP reportó una mutación en el exón 4 c.716G>A, lo que ocasionó un cambio de arginina por histidina en la posición 239 de la proteína (p.Arg239His).
ConclusionesLos signos radiológicos en la resonancia fueron determinantes para el diagnóstico, que posteriormente se confirmó con estudio molecular. Es importante considerar que ciertas mutaciones no se asocian con macrocefalia, lo cual puede ocasionar retraso en el diagnóstico.
Alexander disease is a rare form of leukodystrophy that involves mainly astrocytes; it is inherited in an autosomal recessive manner and occurs by mutations in the GFAP gene, located on chromosome 17q21. It can occur at any age and its infantile form is characterized by macrocephaly, seizures, severe motor and cognitive delay, and progressive spasticity or ataxia.
Case reportAn 8-month-old female was evaluated with a history of neurodevelopmental delay and unprovoked focal motor seizures. Physical examination showed normal head circumference, increased motor responses to tactile and noise stimuli, pyramidal signs and no visceromegalies. Widespread hypodense white matter was found on magnetic resonance and lumbar puncture showed hyperproteinorrachia. Krabbe disease was ruled out by enzymatic assay and gene sequencing of GALC. In the reassessment of the case, abnormalities in neuroimaging lead to suspicion of Alexander disease, and GFAP gene sequencing reported a pathogenic mutation in exon 4 c.716G>A, which caused a change of arginine to histidine at position 239 of the protein (p.Arg239His).
ConclusionsThe radiographic signs observed in the resonance were decisive for the diagnosis, later confirmed by molecular study. It is important to consider that certain mutations are not associated with macrocephaly, which may cause delay in diagnosis.
La enfermedad de Alexander (OMIM #203450) consiste en un desorden neurodegenerativo que forma parte de las leucodistrofias infantiles; es extremadamente rara y afecta principalmente a los astrocitos del hipocampo, el núcleo estriado y la neocorteza1,2. Asimismo, la enfermedad de Alexander se presenta por mutaciones en el gen de la proteína ácida fibrilar glial (GFAP, siglas en inglés), que se localiza en el cromosoma 17q21 y tiene un patrón de herencia autosómica dominante; se observa con mayor frecuencia en mujeres3,4.
Por la edad en que la enfermedad se presenta, se consideran cuatro formas de la misma: neonatal, infantil, juvenil y del adulto5. La forma neonatal inicia en el primer año de vida, es rápidamente progresiva, con crisis convulsivas, hidrocefalia, retraso motor y cognitivo grave y espasticidad o ataxia progresivas. Los pacientes fallecen en los dos primeros años de vida6. La forma infantil es la más frecuente y se presenta en 51% de los casos, generalmente durante los dos primeros años de vida con manifestaciones similares y muerte temprana, aunque algunos pacientes sobreviven hasta la adolescencia. Una de las manifestaciones cardinales de esta variante es la macrocefalia relacionada con megalencefalia7,8. La forma juvenil (23%) inicia entre los cuatro y diez años; los afectados muestran signos bulbares o pseudobulbares como vómitos frecuentes, problemas de lenguaje y deglución, pérdida gradual de funciones cognitivas y el resto de manifestaciones semejantes a las otras formas. La sobrevida es hasta los 20 a 30 años de edad8,9. En la forma adulta (24%), las manifestaciones son muy variables, con signos bulbares o pseudobulbares, piramidales, disfunción cerebelosa, disautonomías, trastornos del sueño y crisis convulsivas10–12.
En este artículo, se describe un caso con enfermedad de Alexander sin macrocefalia y se revisan los principales aspectos del diagnóstico diferencial neuroradiológico y molecular de las leucodistrofias.
2Caso clínicoPaciente de sexo femenino de 8 meses, padres no consanguíneos, producto de la segunda gesta, sin antecedentes perinatales, con contacto visual a los 2 meses, sonrisa social a los 4 meses, no logró sostén cefálico. A los 7 meses presentó crisis convulsivas focales motoras tónico clónicas en hemicuerpo derecho no provocadas. Se inició tratamiento con valproato de magnesio sin mejoría, se agregó clonazepam, y por la persistencia de crisis acudió a esta institución. Durante la exploración física mostró perímetro cefálico normal (p25), adecuado contacto visual, fondo de ojo normal, respuesta motora incrementada ante estímulos táctiles y ruido, hipertonía e hiperreflexia generalizada, reflejo de Babinski y sucedáneos presentes y sin visceromegalias.
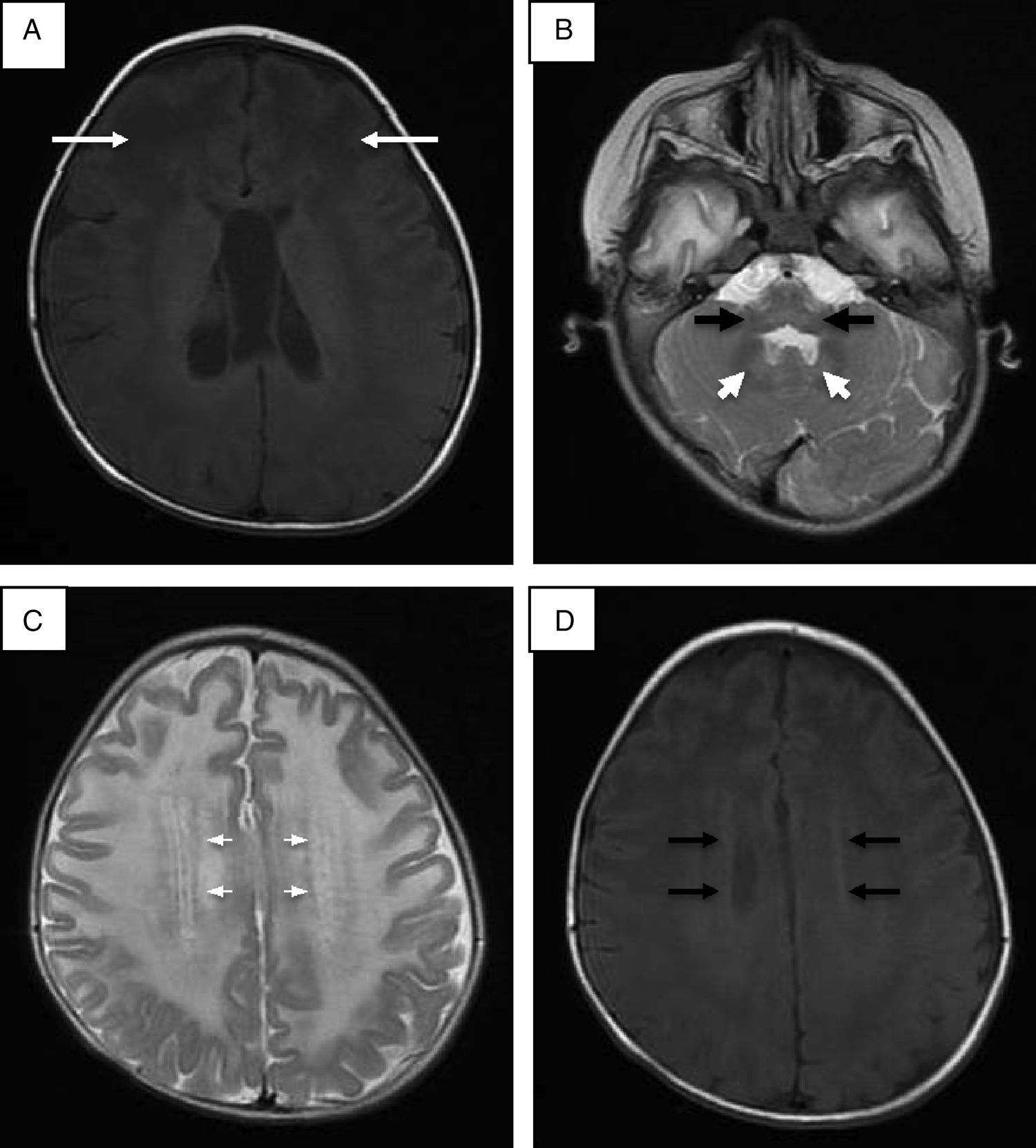
Se realizó punción lumbar, que mostró hiperproteinorraquia; la tomografía de cráneo mostró hipodensidad generalizada de la sustancia blanca. La resonancia magnética (RM) de cráneo corroboró afección de sustancia blanca generalizada, ganglios basales, tálamos, tallo cerebral y cerebelo (fig. 1). Se descartó enfermedad de Krabbe mediante ensayo enzimático y secuenciación del gen GALC. En la reevaluación del caso a los 20 meses de edad, las alteraciones en la RM hicieron sospechar de enfermedad de Alexander, y la secuenciación del gen GFAP reportó una mutación en el exón 4 c.716G>A, que ocasionó un cambio de arginina por histidina en la posición 239 de la proteína (p.Arg239His).

Resonancia magnética de cráneo. A. Imagen potenciada en proyección axial SE en T1 donde las flechas señalan mayor afección de la sustancia blanca de predominio frontal. B. Alteraciones en la señal de la sustancia blanca del cerebelo, núcleo dentado y bulbo raquídeo en imagen potenciada en FSE T2. C. Halo hiperintenso en proyección axial en FSE T2. D. Halo hipointenso en T1 proyección axial.
Las leucodistrofias son un grupo de enfermedades hereditarias que afectan la sustancia blanca del sistema nervioso central y ocasionalmente del sistema nervioso periférico. Abarcan cerca de 40 entidades, y su diagnóstico diferencial en la infancia es complejo, ya que requiere un análisis sistemático que comience con la realización de una adecuada historia clínica y una exploración neurológica completa. Obtener los antecedentes familiares y realizar el árbol genealógico puede orientar hacia padecimientos recesivos ligados al cromosoma X cuando existen tíos maternos afectados o formas autosómicas recesivas en familias con consanguinidad o endogamia13,14.
En la mayoría de los casos hay un periodo de desarrollo normal estático o progresivo (excluyendo las formas neonatales o infantiles tempranas) antes de iniciar la pérdida de habilidades. El inicio de los síntomas en la enfermedad de Alexander puede ocurrir en cualquier etapa de la vida, a diferencia de otras entidades como la enfermedad de Krabbe, que se desarrolla en la infancia temprana8,13.
Las leucodistrofias generalmente no muestran dismorfias, pero en el curso de algunos casos pueden aparecer signos dismórficos. Por ejemplo, en la enfermedad de Canavan y la enfermedad de Alexander se observa macrocefalia, e incluso se considera como rasgo distintivo de estos desórdenes. En la deficiencia del transportador MCT8 de hormonas tiroideas son característicos los trastornos del movimiento como distonía y disquinesias. En la enfermedad de Krabbe la neuropatía periférica es muy frecuente. En adrenoleucodistrofia se presentan leucodistrofia metacromática, bajo rendimiento académico y problemas de atención. En la leucoencefalopatía con sustancia blanca evanescente y en el síndrome de las 4H (hipomielinización con hipogonadismo hipogonadotrópico e hipodontia) predomina la ataxia. Otras entidades desarrollan crisis convulsivas tardías, pero en la enfermedad de Alexander pueden ser la manifestación más temprana13,14. Este caso mostró una variedad de inicio infantil, con retraso en el desarrollo, hipotonía grave y aparición de crisis convulsivas sin macrocefalia.
La revisión y búsqueda de signos en la RM son determinantes para el diagnóstico diferencial de los padecimientos que afectan la sustancia blanca; lo relevante en el estudio de imagen es establecer si se trata de una anormalidad desmielinizante o hipomielinizante según la intensidad de la señal, identificar la región, si afecta tractos específicos y si tiene lesiones confluentes o múltiples. Si el patrón corresponde más a una hipomielinización y ante una entidad lentamente progresiva, se debe considerar síndrome de Allan-Hernon-Dudley por deficiencia del transportador MCT8 de hormonas tiroideas. La ausencia de atrofia cortical es característica de la enfermedad de Pelizaeus-Merzbacher, mientras que la atrofia cerebelosa corresponde al síndrome de 4H. Un compromiso difuso en la sustancia blanca es típico de la leucodistrofia megalencefálica con quistes subcorticales.
Las áreas de afección pueden orientar el diagnóstico: parieto-occipital en adrenoleucodistrofia o enfermedad de Krabbe; temporal en el síndrome de Aicardi-Goutières; subcortical en síndrome de Kearns-Sayre; periventricular en leucodistrofia metacromática y frontal en la enfermedad de Alexander. En la enfermedad de Krabbe se observan hiperintensidades simétricas en el nervio óptico y los tractos piramidales13–15.
Se ha establecido que identificando cuatro de los cinco hallazgos radiológicos característicos de la enfermedad de Alexander se puede tener una mayor certeza en el diagnóstico (tabla 1)16. En este caso se encontraron cuatro criterios; el quinto no fue evaluado por no contar con estudio contrastado. Se ha descrito que las formas adultas y juveniles de la enfermedad de Alexander pueden tener hallazgos poco distintivos, como mayor afección de estructuras de la fosa posterior, atrofia del tallo y cambios difusos de señal en ganglios basales y tálamos. Algunos autores proponen que la afección a estructuras de fosa posterior, atrofia y lesiones tipo tumor en tallo cerebral, anormalidades en la señal en tálamo, núcleos de la base, bulbo o médula espinal, epéndimo empedrado y captación de gadolinio por las lesiones son suficientes para pensar en una forma atípica de enfermedad de Alexander17,18.
Criterios neurorradiológicos de la enfermedad de Alexander
| 1) Afección generalizada de la sustancia blanca de predominio frontal 2) Halo periventricular hipodenso en T2 e hiperintenso en T1 3) Anormalidades en ganglios basales y tálamo, entre las que se incluye edema, hiperintensidad o hipointensidad de señal en secuencia T2 4) Anormalidades en el romboencéfalo, particularmente en bulbo raquídeo y mesencéfalo 5) Reforzamiento con gadolineo del halo periventricular, sustancia blanca frontal, fornix, quiasma óptico, núcleos de la base, tálamos, núcleo dentado y tallo cerebral |
Antes del diagnóstico molecular, la presencia de depósitos proteicos (denominados fibras de Rosenthal) en una biopsia cerebral constituía la única forma de confirmar la enfermedad de Alexander. Durante el curso del padecimiento, estas fibras (compuestas por agregados de proteína ácida fibrilar glial (GFAP), vimentina, αβ-cristalina y la proteína de choque térmico 27) van aumentando en tamaño y número. La GFAP es un componente del citoesqueleto de 432 aminoácidos que provee estabilidad estructural al astrocito maduro19–21.
Se han descrito más de 100 mutaciones en el gen de la GFAP que afectan a la proteína con un efecto dominante negativo; es decir, la proteína mutante impide la función de la proteína silvestre a través de un mecanismo tóxico que no está del todo entendido2. Se postula que no hay un efecto directo sobre la síntesis de la proteína, sino que la proteína mutante afecta el proceso de oligomerización o solubilidad. La oligomerización anormal también podría inhibir la actividad del proteasoma en el astrocito, de tal manera que se afecta la interación normal entre astrocitos y oligodendrocitos, resultando en hipomielinización o desmielinización. Se ha demostrado mediante inmunohistoquímica una disminución en los niveles del transportador 1 del glutamato en los astrocitos del hipocampo. Asimismo, diferentes estudios de expresión han evidenciado que la disminución en la captura del glutamato tiene un rol muy importante en el daño neuronal. El exceso de glutamato extracelular incrementa la actividad excitatoria neuronal y se manifiesta como crisis convulsivas de muy difícil control22.
La mayoría de las variantes genéticas de la enfermedad de Alexander corresponden a mutaciones con sentido erróneo (en especial Arg79, Arg88, y Arg239), y en conjunto se encuentran en 34% de los casos. Las variantes patogénicas más recurrentes exclusivas de las formas infantil, juvenil y adulta se presentan en la tabla 2. Algunas mutaciones, como la p.Arg416Trp, se han descrito en todas las formas de la enfermedad8.
Mutaciones patológicas específicas de cada forma
| Forma infantil | Forma juvenil | Forma Adulto |
|---|---|---|
| p.Met73Thr | p.Leu235Pro | p.Arg66Gln |
| p.Leu76Phe | p.Arg70Trp | |
| p.Asn77Ser | p.Arg70Gln | |
| p.Lys86_Val87delinsGluPhe | p.Met74Thr | |
| p.Leu97Pro | p.Glu205Lys | |
| p.Arg239His | p.Leu359Pro | |
| p.Arg239Leu | p.Ala364Thr | |
| p.Leu352Pro | ||
| p.Glu373Lys | ||
| p.Asp417MetfsTer15 |
En el presente caso se demostró una mutación en el exón 4 (c.716G>A) que afecta el dominio enrollado 2A de la proteína. Las mutaciones son más frecuentes en el exón 1 (45%), seguido por el 4 (27.2%) y el 6 (16%). De acuerdo con Prust y colaboradores, en un estudio de 215 casos no se encontraron mutaciones en los exones 2 y 95. Los autores proponen la existencia de dos fenotipos basados en la edad de inicio: el tipo 1 o cerebral, de inicio temprano, y el tipo 2 o bulbar, de inicio tardío. El tipo 1 incluye los casos más típicos con macrocefalia, detención del crecimiento, encefalopatía, crisis convulsivas, retraso cognitivo y motor en los primeros 4 años de vida, además de presentar los hallazgos radiológicos clásicos descritos por van der Knaap y colaboradores16. El tipo 2 muestra un cuadro atípico en el que predominan signos bulbares, disfunción autonómica, signos cerebelosos, disfonía, anormalidades en los movimientos oculares y mioclono palatino; en este fenotipo predominan las anormalidades de la sustancia blanca de la fosa posterior, del tallo cerebral, del cerebelo y la atrofia de médula espinal.
En el caso aquí presentado, el residuo de arginina se localiza en la posición 239, que resulta ser un sitio frecuente de mutación (20.3% de los casos), además de ser altamente conservado entre especies. Los casos clínicos que portan esta mutación tienen el fenotipo cerebral de la enfermedad y la mayoría presentan macrocefalia. En la enfermedad de Alexander, este trastorno suele desarrollarse rápidamente tal vez como resultado de la proliferación de astrocitos atípicos. Es posible que los casos sin macrocefalia tengan una disrupción del equilibrio energético celular que evita la proliferación neuronal. En la tabla 3 se comparan casos con afección en Arg239 que muestran edad de inicio temprano y retraso psicomotor5. Es evidente que esta paciente no mostró macrocefalia o neuroregresión.
Comparativo de casos con afección en Arg239
| Goyal et al23. | Caroli et al24. | Nuestro caso | |
|---|---|---|---|
| Edad diagnóstico | 16 meses | 4 meses | 8 meses |
| Crisis convulsivas (edad de presentación) | + (4 meses) | + (NR) | + (7 meses) |
| Retraso psicomotor | + | NR | + |
| Regresión | + | NR | - |
| Espasticidad | + | + | + |
| Hiperreflexia | + | + | + |
| Macrocefalia | - | + | - |
| Hallazgos RMa | 1,2,3 | 1,2,3,4 | 1,2,3,4 |
NR: no reportado; RM: resonancia magnética.
La enfermedad de Alexander es un desorden que muestra manifestaciones clásicas que son inconfundibles; sin embargo, es importante tenerla en consideración como diagnóstico diferencial ante cuadros atípicos sin macrocefalia o sin neuroregresión. Hay datos en la RM que pueden ser útiles en el diagnóstico diferencial con otras leucodistrofias, sobre todo cuando el inicio de las manifestaciones es temprano, aun cuando la macrocefalia no esté presente.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores han obtenido el consentimiento informado de los pacientes y/o sujetos referidos en el artículo. Este documento obra en poder del autor de correspondencia.
FinanciamientoNinguno.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
A la Dra. Merjo van der Knaap por sus valiosas aportaciones durante el análisis clínico y radiológico de la paciente.