



Introducción: La enfermedad de Pompe (EP) es un trastorno autosómico recesivo causado por deficiencia de la enzima lisosomal, α-glucosidasa ácida (GAA) humana. Este déficit produce depósitos de glucógeno en hígado y músculos. La EP en niños pequeños (inicio temprano) produce cardiomiopatía e hipotonía. Sin embargo, en el inicio tardío, la cardiomiopatía se considera poco frecuente, y se ha dicho que hay menor beneficio con la terapia de reemplazo enzimático (TRE) con GAA.
Caso clínico: Paciente femenino de 8 años, que inició con síntomas a los 3 años de edad (inicio tardío) con infecciones respiratorias recurrentes y debilidad muscular progresiva. Se diagnosticó con EP por evidencia en biopsia muscular y la baja actividad de α-glucosidasa. Se realizó lobectomía derecha basal por bronquiectasias y necrosis. Desarrolló neumonía y recibió asistencia respiratoria mecánica (CPAP) por 4 semanas, con dependencia absoluta de oxígeno y BiPAP. Se aplicó TRE con GAA.
Conclusiones: La evolución positiva de la paciente, especialmente en las funciones respiratoria y cardiovascular, muestra que es posible mejorar la cardiomiopatía en pacientes con EP de inicio tardío.
Background: Pompe disease (PD) is an autosomal recessive disease caused by a deficiency in the lysosomal human enzyme α-alglucosidase. Among children (early onset), PD causes cardiomyopathy, whereas late-onset disease seems unrelated to a high rate of cardiomyopathy. Patients respond less to enzyme replacement therapy (ERT) with α-alglucosidase.
Case report: This is the case of an 8-year-old female patient with symptom onset at 3 years of age (late onset) with recurrent respiratory infections and progressive muscular weakness. Diagnosis for Pompe disease (PD) was established due to evidence from muscle biopsy. At baseline, a right lobectomy was performed for bronchiectasis and necrosis. The patient developed pneumonia and received mechanical respiratory support (CPAP) for 4 weeks with absolute dependence on oxygen and BPAP. ERT with α-alglucosidase was given.
Conclusions: This patient's positive output and remarkable effects on cardiovascular and respiratory function suggest that ERT may reduce cardiomyopathy among late-onset PD patients.
Pagina nueva 1
Introducción
Las enfermedades por depósito lisosomal son un grupo de errores congénitos del metabolismo que pueden ser de origen genético autosómico recesivo o recesivo ligado al cromosoma X. El efecto es un déficit enzimático que permite la acumulación anormal del sustrato no degradado, y produce daño y manifestaciones clínicas específicas en diversos órganos.
Son consideradas raras y exhiben muy bajas tasas de incidencia, pero son un problema de salud pública por causar muerte prematura, tener un curso crónico, ocasionar trastornos neurológicos importantes y deterioro de la calidad de vida1,2.
Hasta hace 20 años solo se brindaba tratamiento de las complicaciones, por lo que la conducta era expectante, y no se podía cambiar su curso. Actualmente, con la emergencia de la terapia de reemplazo enzimático se abre una puerta a la posibilidad de cambiar el curso de este grupo de enfermedades.
El déficit enzimático hace de la terapia de reemplazo enzimático (TRE) el principal recurso terapéutico para el manejo clínico. Un ejemplo de su eficacia se registró en la enfermedad de Gaucher (EG). Hace dos décadas tan solo, la única terapia existente eran paliativos para el manejo de las complicaciones. Al día de hoy, la TRE es hoy un hito para el tratamiento de la EG por su capacidad de controlar la sintomatología, disminuir la discapacidad y mejorar la calidad de vida1,2.
Enfermedad de Pompe
La Enfermedad de Pompe (EP) fue descrita por el patólogo holandés Joannes C. Pompe. También se le conoce como glucogenosis tipo II (GSD II). En 1932, Pompe publicó el caso clínico de una paciente de 7 meses de edad con cardiomegalia, que falleció por aparentes complicaciones de neumonía, cuya necropsia mostró el depósito masivo de glucógeno, afectando principalmente al músculo esquelético y al músculo cardiaco. Tres décadas después (1963), Herz observó las vacuolas con depósito de glucógeno por microscopía electrónica, y describió la enfermedad por depósito lisosomal. Ante estos hallazgos, se ha consignado que la afección muscular es secundaria al depósito de glucógeno en los lisosomas por déficit en la enzima α-glucosidasa ácida (GAA)3.
Manifestaciones clínicas
Las manifestaciones clínicas cardinales de EP son debilidad e hipotonía, por lo que se considera también una enfermedad neuromuscular o miopatía metabólica. Su variabilidad fenotípica ha llevado a clasificarla en dos tipos, según la edad de presentación: EP de inicio temprano y de inicio tardío. Actualmente, la EP se considera un espectro continuo de manifestaciones que varían según la actividad residual de GAA, y que conduce, invariablemente, a debilidad progresiva y muerte por acumulación de glucógeno3,4.
En promedio, la EP de inicio temprano comienza a los 2 meses, el diagnóstico se realiza a los 4 meses, el tiempo para un primer soporte ventilatorio es de 5.9 meses y la supervivencia de 8.7 meses. Sin tratamiento, la EP exhibe una mortalidad promedio de 74% al año de edad y de 88% a los 18 meses. La muerte usualmente se debe a insuficiencia cardiaca, con hipotonía generalizada, y debilidad de músculos respiratorios. A nivel cardiaco se registra engrosamiento progresivo del ventrículo izquierdo (VI) por deterioro de la función sistólica y el desarrollo de insuficiencia cardiaca progresiva, que causa la muerte antes del primer año de vida. Diversos autores consideran la EP de inicio temprano como EP infantil, y la enfermedad de inicio tardío como EP juvenil3,5,6.
Un estudio realizado en una serie de 19 pacientes con EP infantil o de inicio temprano mostró un intervalo PR corto en hasta el 75% de los casos, así como que la interferencia con tejidos de conducción promueve complejos QRS muy altos en los electrocardiogramas. Estas alteraciones en la conducción cardiaca y la hipertrofia ventricular aumentan el riesgo de presentar taquiarritmias de manera súbita, lo que puede ser fatal en situaciones de incremento del gasto cardiaco. El intelecto de los pacientes siempre es normal5,6.
Terapia de reemplazo enzimático en la enfermedad de Pompe
La terapia de reemplazo enzimático (TRE) está disponible desde el año 2000, gracias a la tecnología recombinante de ADN. En 2007, un trabajo con 18 pacientes menores de 6 meses de edad con EP y cardiomiopatía, tratados con TRE a base de GAA durante 52 semanas, mostró que hubo reducción en la utilización de ventilación asistida y en la mortalidad. Trabajos clínicos de años posteriores han demostrado que la TRE mejora sustancialmente las tasas de supervivencia y la función de músculos respiratorios, así como la reducción de la incapacidad por debilidad en el músculo esquelético5,7.
En EP de inicio temprano, la TRE ha demostrado incidir en la remodelación miocárdica, mejorar la función ventricular y disminuir el grosor de la pared. En EP de inicio tardío, la afección cardiaca es menos frecuente, por lo que se estima la eficacia de la TRE como limitada, sin literatura de una mejoría significativa en la función y el tamaño ventricular8.
Por los avances logrados con TRE en las enfermedades por depósito lisosomal, es importante determinar si existen o no cambios significativos en la remodelación y función miocárdica en pacientes con EP de inicio tardío. Esta experiencia con administración de TRE en una niña mexicana con EP de inicio tardío con una muy buena respuesta y remisión del daño cardiaco es, desde esta perspectiva, un caso excepcional, que debe consignarse en la literatura nacional e internacional9-11.
Con base en lo anterior, el objetivo de este trabajo fue describir la evolución de la función miocárdica en una paciente mexicana, evaluando beneficio de reversión de la miocardiopatía con TRE y comparando los resultados de la función miocárdica con TRE de inicio tardío con la literatura a nivel internacional. Además, se buscó describir puntualmente la evolución de la función músculoesquelética, incluida la función respiratoria, y la evolución de la misma a partir del inicio de TRE.
Caso clínico
Se presenta el caso de una paciente de sexo femenino, de 9 años de edad, originaria del Estado de México, que ingresó al Hospital Infantil de México Federico Gómez en el 2011, con antecedentes de padres sanos, sin consanguinidad o endogamia. Proviene de nivel socioeconómico bajo, del medio rural, sin antecedentes perinatales importantes, con desarrollo psicomotor adecuado para la edad.
Como antecedentes personales patológicos presentó infecciones recurrentes de vías aéreas, diagnosticadas como asma a los 3 años. Cursó con 4 episodios graves de neumonía. Al ingresar presentó ortopnea de 4 años de evolución, que incrementó durante el último año. La paciente requería de 2 a 3 almohadas para dormir, y se presentó con disnea y tos de 4 años de evolución. Se refirieron caídas frecuentes desde su propia altura, de 6 meses de evolución, recurrencia de los cuadros de infección respiratoria, edema progresivo en cara y cianosis peribucal.
Ingresó a urgencias con un cuadro de neumonía y derrame pleural, insuficiencia cardiaca, debilidad muscular marcada. Deambulaba con ayuda.
En los exámenes de laboratorio de marzo del 2011, la biometría hemática y los electrolitos séricos estuvieron dentro de los parámetros normales. La prueba de función hepática mostró cifras de deshidrogenasa láctica de 688, bilirrubina total de 0.75, alanino aminotransferasa de 217 y aspartato aminotransferasa de 511. En la gasometría arterial se registró pH 7.39, pCO2 de 46, pO2 de 58, sat de 89%, y HCO3 de 26.6.
Pruebas de imagen
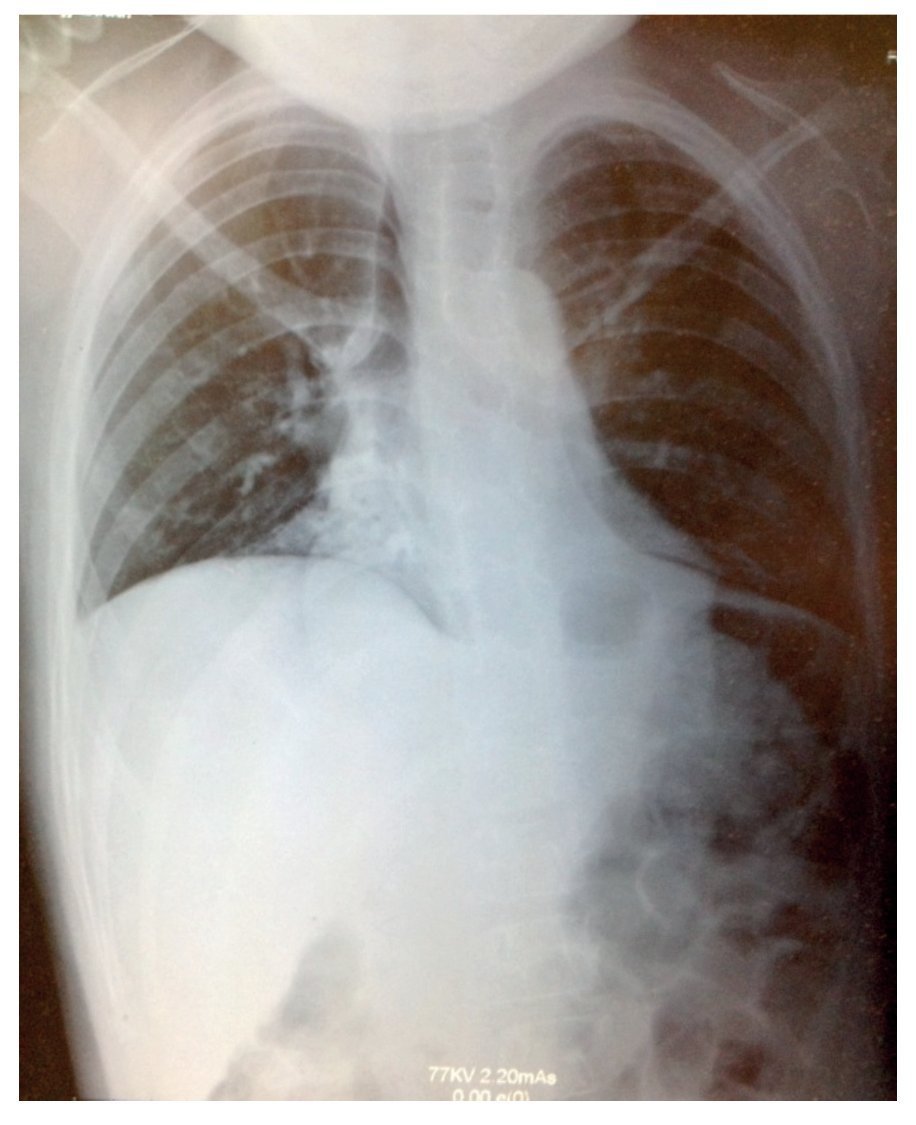
La radiografía simple mostró datos compatibles con neumonía: tórax con infiltrado micronodular derecho de predominio basal (fig. 1). Al realizarse tomografía de tórax con ventana pulmonar, esta mostró una imagen sugestiva de consolidación bilateral, basal y medial con broncograma aéreo, así como derrame pleural derecho sin compromiso de la función (<10%).
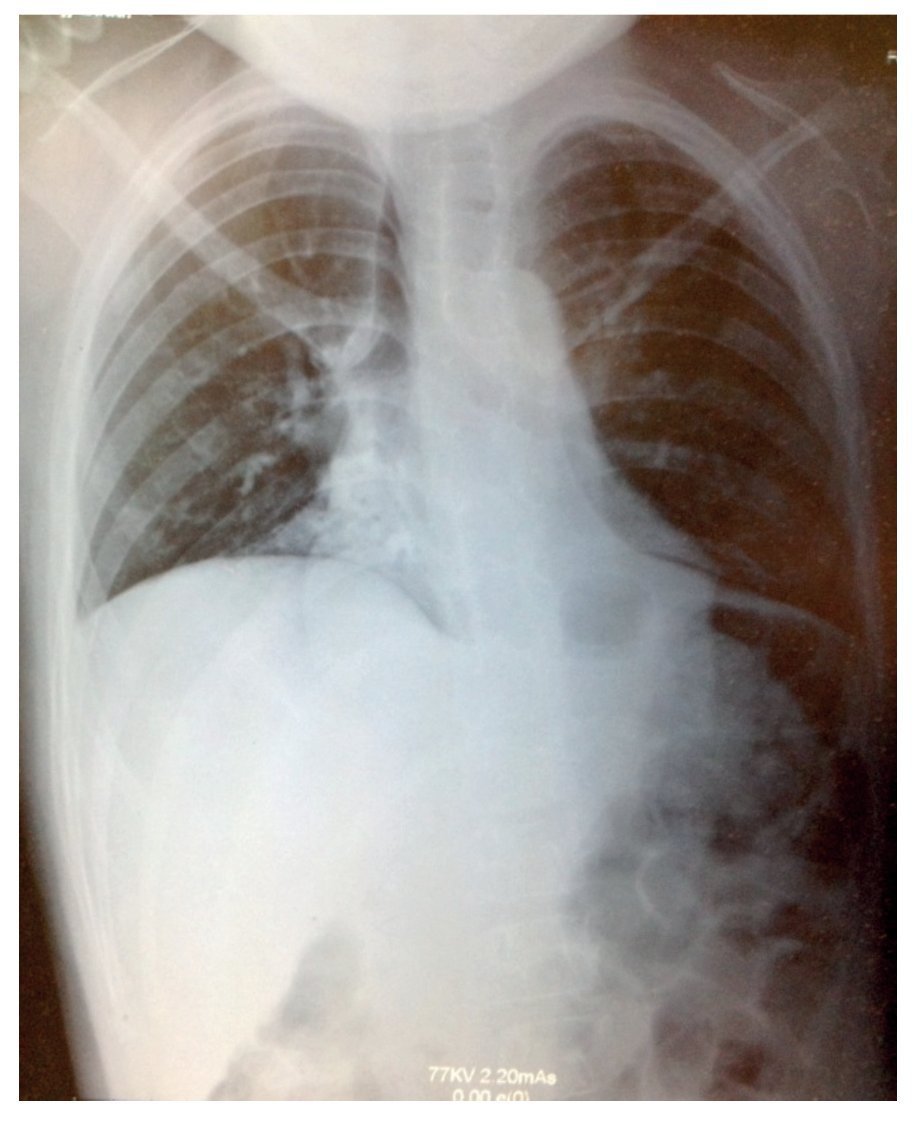
Figura 1 Radiografía con datos compatibles de neumonía, y tórax con infiltrado micronodular derecho de predominio basal.
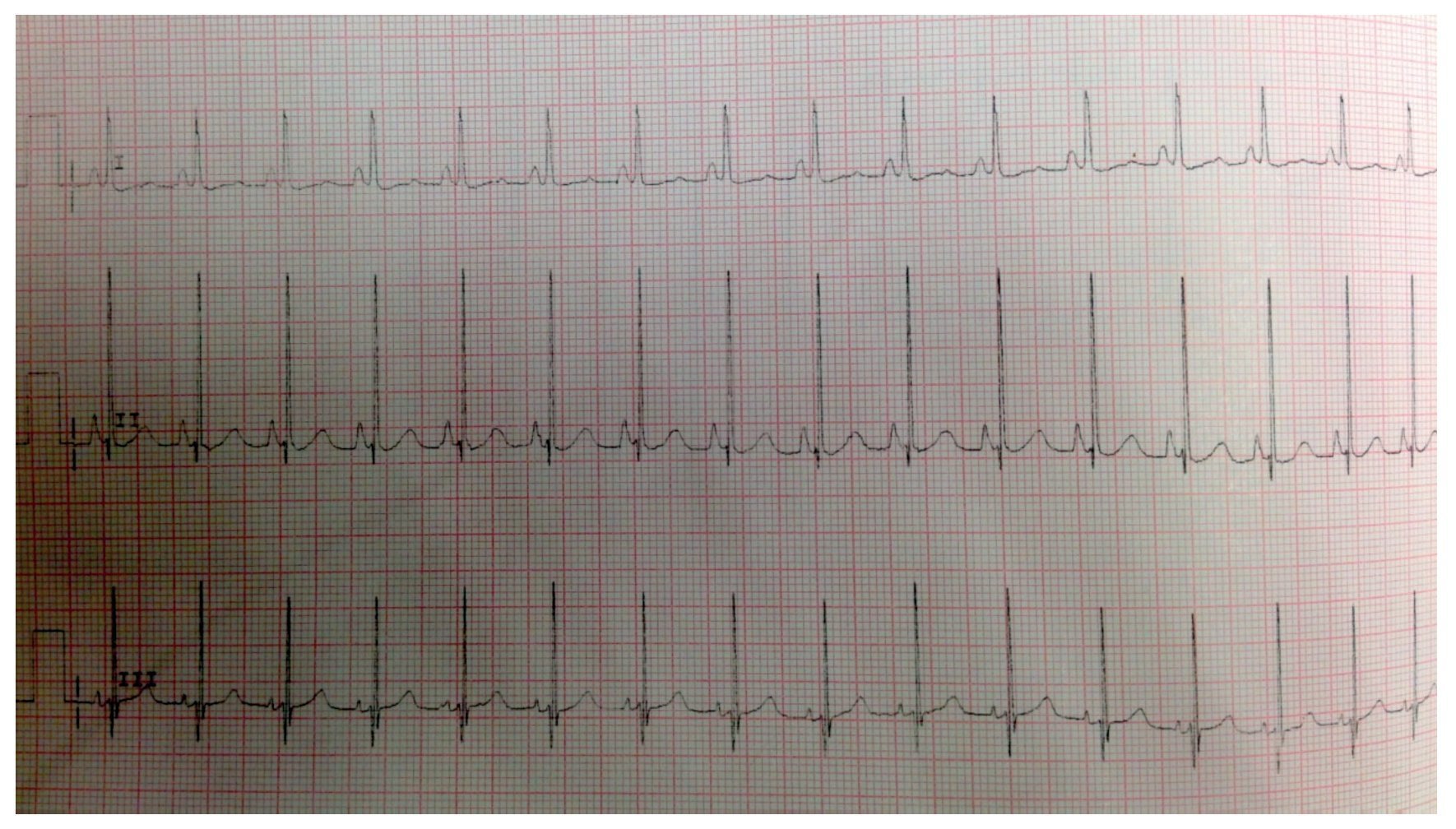
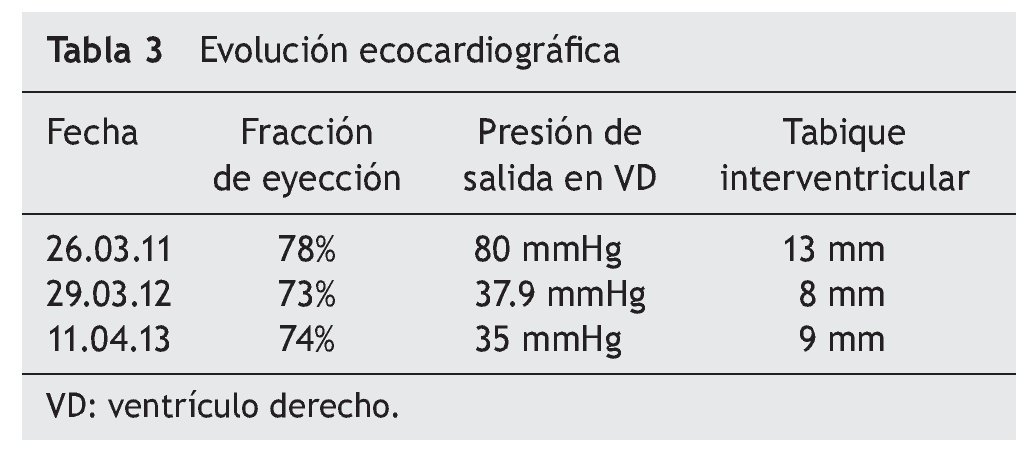
El electrocardiograma mostró ritmo sinusal, hipertrofia biventricular, crecimiento auricular derecho y trastorno en la repolarización ventricular (fig. 2). Se realizó ecocardiograma transtorácico (ETT) que mostró análisis segmentario normal para situs solitus sin defectos estructurales, función ventricular normal con fracción de eyección (FE) del 78%. La presión sistólica de ventrículo derecho (PSVD) se estimó con base en regurgitación de la válvula tricúspide de 80 mmHg. También se observó dilatación cualitativa de cavidades derechas, tronco de arteria pulmonar y ramas.
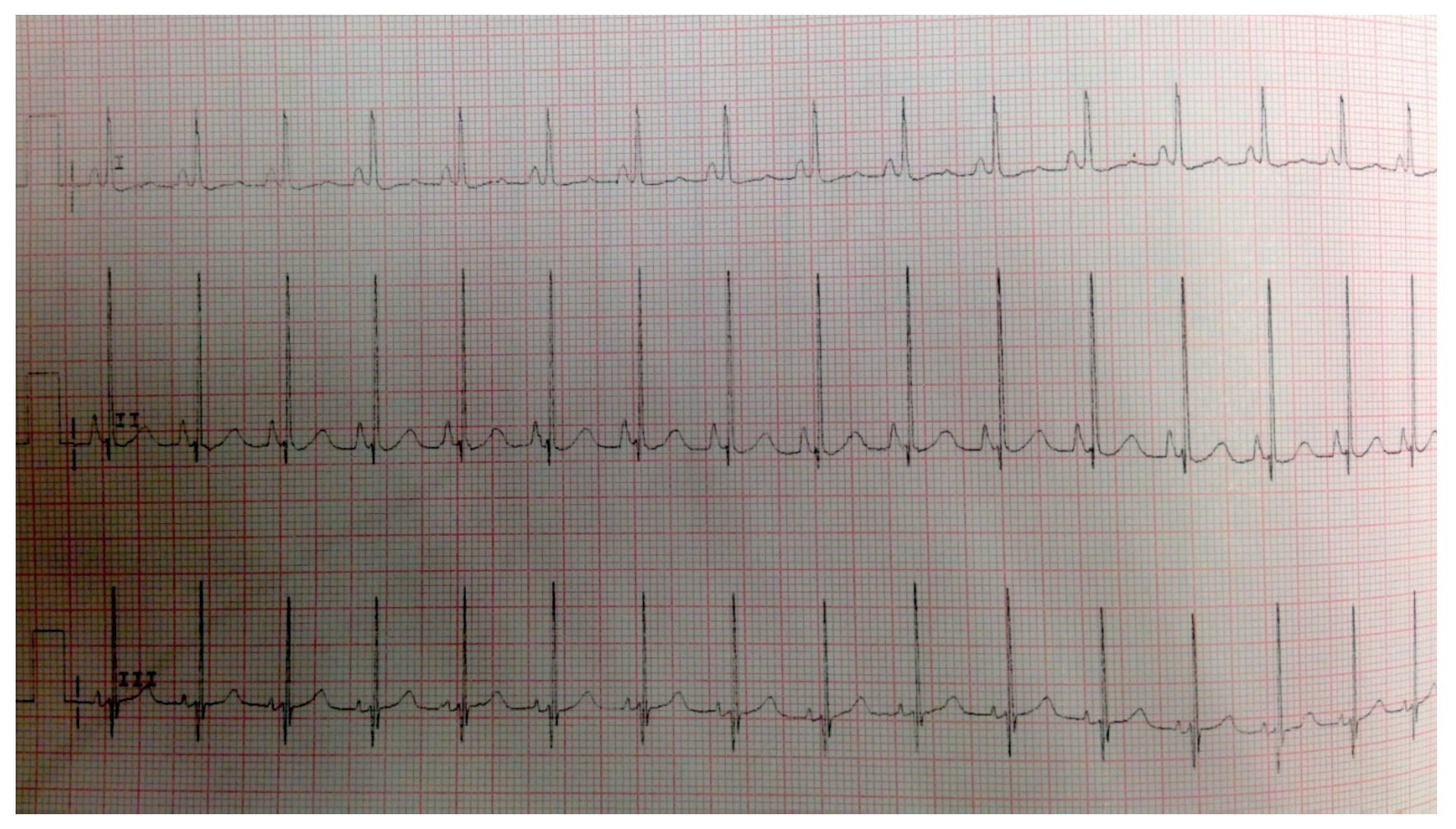
Figura 2 Electrocardiograma con segmento ST aplanado, ondas Q DIII, AVF con inversión de onda T que indica sobrecarga derecha.
Los hallazgos de neumopatía crónica llevaron al abordaje clínico para descartar causas frecuentes, como inmunodeficiencia primaria, fibrosis quística, enfermedad por reflujo gastroesofágico e infección por micobacterias. Algunos de los resultados se presentan a continuación.
• Espirometría: atrapamiento de aire de leve a moderado, poco valorable por uso de presión positiva, con restricción importante.
• Broncoscopia y lavado bronquioalveolar: hipofaringe, glotis y aritenoides sin alteraciones, con tráquea y bronquios de aspecto normal.
• BAAR seriado: negativo.
• Sudán III: negativo.
• Formas invasivas en broncoaspirado con blastoconidias y pseudomicelios sin correlación clínica.
• Estudio de ELISA: negativo.
• Inmunoglobulinas y complemento: normales para la edad.
• Serie esófago-gastro-duodenal: sin alteraciones.
• Electrolitos en sudor: Cl 11.6 mEq, Na 9.3 mEq, K 2 mEq, descartando a la fibrosis quística.
• Gammagrama perfusorio-ventilatorio: datos asociados con hipertensión pulmonar y patología mixta. Bronquiectasias cilíndricas bibasales.
• Lobectomía basal: lóbulo inferior izquierdo con poca fibrosis, cambios macroscópicos del parénquima pulmonar, lóbulo superior y língula de características normales.
En el posquirúrgico, presentó hipoventilación alveolar y acidosis respiratoria. Se requirió soporte ventilatorio mecánico por cuatro días, con 23 días de estancia total en terapia intensiva. Cursó con crisis convulsiva secundaria a hipercapnia (pCO2 139). Se aplicó ventilación con BiPAP (sistema de bipresión positiva), descartando fibrosis quística. En pruebas de linfocitos por cartometría de flujo, se observaron los anticuerpos comunes de una enfermedad autoinmune, complemento, ANA, Anti DNA, pANCA y cANCA. Se documentó complemento y cartometría de flujo dentro de parámetros normales para la edad.
Ante la debilidad muscular progresiva, se indicó serología para Trichinella spiralis que resultó positiva; sin embargo, el hallazgo se desestimó al no correlacionar con afectación cardiaca y el resultado de CPK. Se continuó el abordaje de miopatía. El estudio de electromiografía para neuroconducción motora y sensorial de las cuatro extremidades mostró parámetros normales, pero el estudio de fibra muscular fue anormal, mostrando cambios de tipo inflamatorio. Se realizó polisomnografía ante la sospecha de apnea obstructiva, confirmándose por retención de CO2 e hipoxia.
Al realizarse la biopsia muscular (cuádriceps) se hallaron inclusiones citoplasmáticas masivas de glucógeno. Se concluye que se trata de glucogenosis tipo II o enfermedad de Pompe. El diagnóstico se confirmó con la determinación de actividad enzimática. Los valores obtenidos fueron los siguientes.
• Alfa glucosidasa con pH 3.8: 0.24 (valores de referencia 1.5-10 nmol/spot de 21 h).
• Alfa glucosidasa con pH 7.0: 1.59 (valores de referencia 1.8-17.1 nmol/spot de 21 h).
• Alfa glucosidasa con inhibición: 0.03 (valores de referencia 0.9-7.2 nmol/spot de 21 h).
El resultado de la secuenciación de ADN confirmó dos mutaciones descritas para enfermedad de Pompe: C.258dupC (heterocigoto) y C.1445C>T (heterocigoto). Antes de iniciar con la TRE, la paciente cursó con dos episodios de neumonía y un paro cardiorrespiratorio. Requirió reanimación avanzada, intubación y ventilación mecánica por 4 semanas. Después de varios intentos fallidos, se logró la extubación; se continuó con soporte en modo BiPAP durante el día y durante el sueño, alternando con aporte de oxígeno en puntas nasales. Recibió nutrición parenteral y gastrostomía por incapacidad de deglutir, por la debilidad muscular y el compromiso del estado nutricional.
El 20 de febrero del 2012 se decidió iniciar con TRE, administrando una infusión de α-glucosidasa ácida (20 mg/kg/ cada 2 semanas).
Evolución de la terapia de reemplazo enzimático durante el primer año de tratamiento
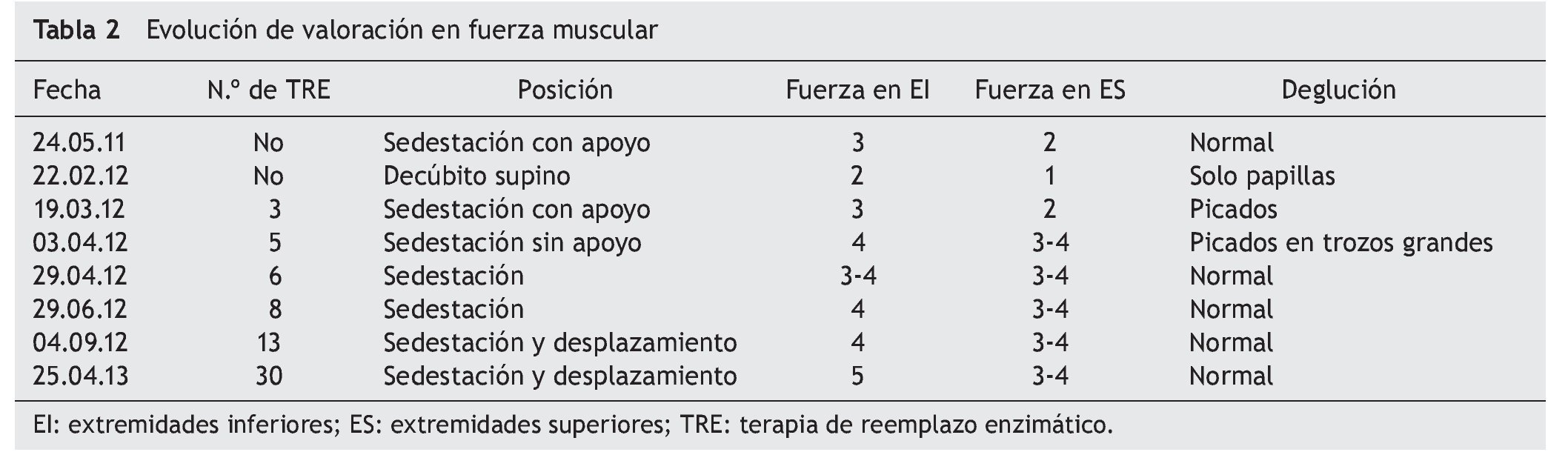
Al inicio de TRE, la valoración de rehabilitación mostró una deficiencia músculo-esquelética severa que limitaba la realización de actividades cotidianas y la participación social. Se apreciaron arcos de movilidad pasivos completos con fuerza de 2/5, en su mayoría en músculos del tronco y extremidades superiores, y de 1/5 en extremidades inferiores, según la escala de Daniels (tabla 1).

Una semana después de la primera infusión enzimática se registró progreso del modo ventilatorio, pasando de SIMV a CPAP, según tolerancia. Mejoró, además, la deglución, pasando a dieta normal blanda, que fue parcialmente tolerada. Y hacia finales del primer mes, se registró mejoría en fuerza muscular, aunque el esfuerzo respiratorio era aún insuficiente para retirar la ventilación asistida. Hacia las seis semanas, se cambió la ventilación a modalidad BiPAP.
A los dos meses, se registró una mejoría notable en cuanto al sostén de tronco, sedestación y movilidad en extremidades inferiores, de acuerdo con la escala de Daniels, con fuerza muscular intrínseca y prono supinadores de 3; tríceps, bíceps, flexores y abductores de hombros de 2; flexores de cadera de 3, y extensores de rodilla, eversores, flexores y extensores de tobillo de 1 (tabla 2).
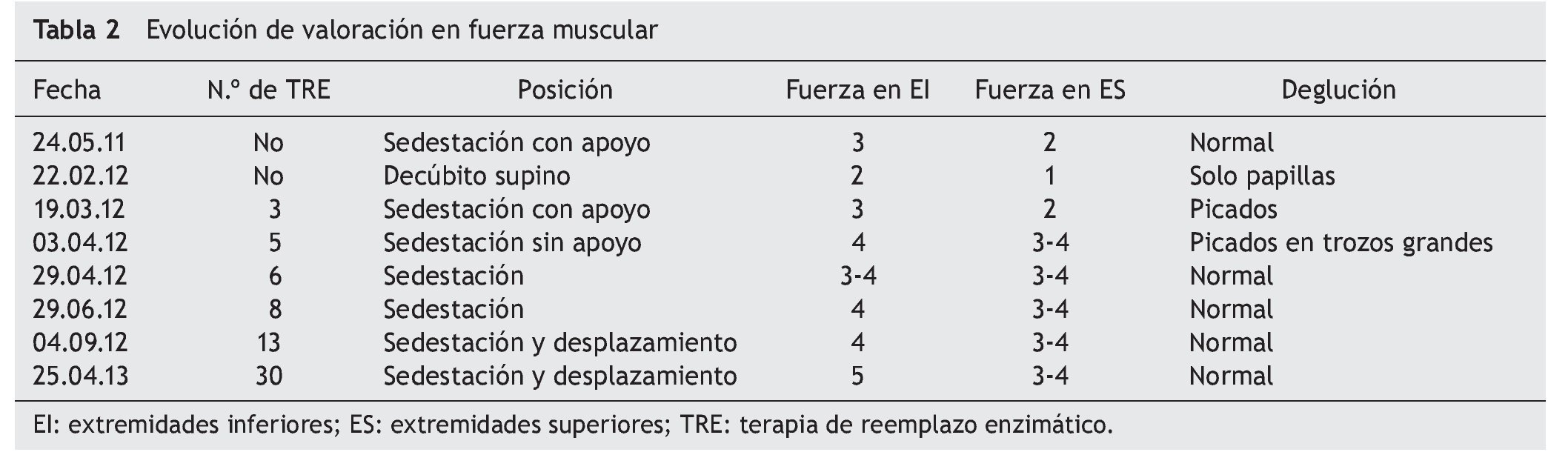
Hacia el tercer mes de TRE se logró el retiro de BiPAP intermitente, con oxígeno en puntas nasales. La paciente egresó para continuar con manejo ambulatorio, con tratamiento de hipertensión pulmonar con anticongestivo y vasodilatador pulmonar, así como manejo ventilatorio con BiPAP intermitente. La paciente tenía equilibrio de cuello y tronco, así como apoyo de manos. Evolucionó de posición acostada a sentada, con dificultad, desplazándose distancias cortas con arrastre, e iniciando alimentación independiente. Miembros superiores e inferiores aún limitados a la extensión de rodilla con regazo de 30º. La fuerza muscular fue evaluada con escala de Daniels (tabla 1) en miembros superiores (bíceps, prono-supinadores y mano en 3, resto 2, fuerza en miembros inferiores en 2).
Hacia el quinto mes mejoró la movilidad, con desplazamiento gateando y con arrastre. El ecocardiograma registró franca mejoría en grosor de septum y pared ventricular. Al finalizar el semestre de TRE, hubo clara mejoría en la postura de la espalda, mejor esfuerzo respiratorio y ausencia de fatiga. Se intercalaron 2 horas de oxígeno inhalado con 2 horas de BiPAP. La paciente mostró mejor tono muscular en las piernas. Se alternaron 3 horas de BiPAP con 3 horas de oxígeno con puntas nasales. Dormía con BiPAP. Ante la evidencia de mejoría de la hipertensión pulmonar se decidió suspender el tratamiento con sildenafil y furosemida, vigilando de forma estrecha cualquier dato de recaída.
Al concluir el primer semestre con TRE, la paciente presentó datos clínicos de pubertad precoz (hirsutismo, botón mamario y vello axilar), por lo que se valoró perfil hormonal, que se encontró normal.
Ocho meses después solamente utilizaba BiPAP durante el sueño y se iba suspendiendo el oxígeno por puntas nasales. La gasometría mostró pH de 7.38, CO2 de 36, O2 de 144. Se observó mejoría en estado ventilatorio con disminución en la retención de CO2.
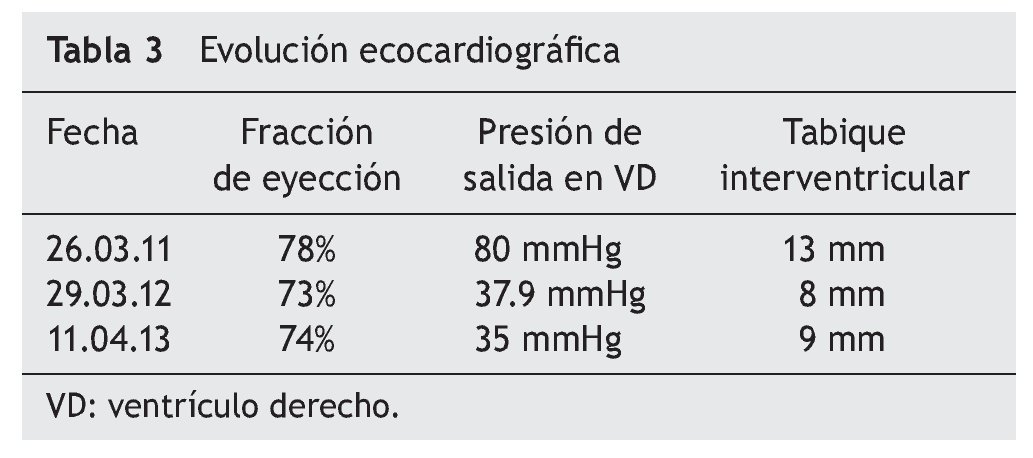
Al cumplirse el primer año de TRE se realizó ecocardiograma que mostró disminución del grosor del septum interventricular (8 mm), PSVD de 35 mmHg y función sistólica del mismo VD conservada, con fracción de eyección del 74% y fracción de acortamiento del 42% (tabla 3).
Actualmente, la paciente se encuentra en rehabilitación y la mejoría en la función motora es evidente; además, acude a la escuela con adecuado aprovechamiento. En general, la calidad de vida de la paciente ha mejorado considerablemente.
Discusión
La EP tiene un amplio espectro clínico al derivar del depósito de glucógeno en tejido muscular y visceral. Es evidente que la función cardiaca en la variante de inicio temprano presenta un extremo compromiso, pero se pueden obtener resultados satisfactorios con TRE, logrando una remodelación del miocardio y de la función ventricular, así como disminución del grosor de la pared ventricular. En EP tardía, por lo general, se describe la afección cardiaca como poco frecuente o menos grave, y los resultados de TRE en relación con la función cardiaca han sido desalentadores y no se ha descrito mejoría en la función o en el tamaño ventricular. En este caso clínico existe evidente afectación del músculo cardiaco, así como insuficiencia cardiaca al inicio de la enfermedad (como se mostró en el ecocardiograma) con hipertrofia de ventrículo derecho e hipertensión pulmonar, documentándose además presión de salida del VD de hasta 80 mmHg. A diferencia de los informes que existen en la literatura, donde no se registra un cambio significativo en la función ventricular o en el grosor de la pared entre pacientes con EP tardía, la evolución de esta paciente demostró que la TRE puede lograr una disminución significativa de la presión de salida de VD, de 80 mmHg hasta 35 mmHg. Asimismo, se documenta una disminución en el grosor inicial del tabique interventricular, de 13 mm a 9 mm. Con la reversión de la insuficiencia cardiaca, se suspendió el tratamiento anticongestivo y vasodilatador.
La EP es una enfermedad que, por su baja incidencia y su amplio espectro de presentación clínica, comprende un reto diagnóstico para el pediatra. La importancia del diagnóstico temprano se asocia con el inicio oportuno del tratamiento específico para evitar la progresión de la enfermedad hacia una incapacidad de los músculos esquelético, respiratorio, cardiaco y muerte, así como para fomentar una mejor calidad de vida.
Debido a los pocos casos documentados y al descubrimiento relativamente reciente de la TRE, su espectro de acción aún no es completamente conocido. Como se demuestra en este caso clínico, parece posible lograr la remodelación cardiaca con mejoría en la función ventricular, aun en pacientes con EP de inicio tardío. Son necesarias más evidencias clínicas de la evolución de la cardiomiopatía con el uso de TRE en pacientes con EP tardía. Es posible integrar de nuevo a la vida normal a los pacientes con EP con tratamiento basado en TRE.
El estado general de la paciente mejoró. Gradualmente, la paciente recuperó 13 kg y 12 cm en 18 meses de TRE. Su actividad física se incrementó, acude a la escuela y su calidad de vida mejoró considerablemente.
Conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
Recibido el 22 de octubre de 2013;
aceptado el 12 de diciembre de 2013
* Autor para correspondencia.
Correo electrónico: magyceron@yahoo.com (M. Cerón-Rodríguez).