


Fifty years after the first reports of Epstein-Barr virus (EBV)-associated endemic Burkitt's lymphoma, EBV has emerged as the third most prevalent oncogenic virus worldwide. EBV infection is associated with various malignancies including Hodgkin and non-Hodgkin lymphoma, NK/T-cell lymphoma and nasopharyngeal carcinoma. Despite the highly specific immunologic control in the immunocompetent host, EBV can cause severe complications in the immunocompromised host (namely, post-transplant lymphoproliferative disease). This is particularly a problem in patients with delayed immune reconstitution post-hematopoietic stem cell transplant or solid organ transplant. Despite advances in diagnostic techniques and treatment algorithms allowing earlier identification and treatment of patients at highest risk, mortality rates remain as high as 90% if not treated early. The cornerstones of treatment include reduction in immunosuppression and in vivo B cell depletion with an anti-CD20 monoclonal antibody. However, these treatment modalities are not always feasible due to graft rejection, emergence of graft vs. host disease, and toxicity. Newer treatment modalities include the use of adoptive T cell therapy, which has shown promising results in various EBV-related malignancies. In this article we will review recent advances in risk factors, diagnosis and management of EBV-associated malignancies, particularly post-transplant lymphoproliferative disease. We will also discuss new and innovative treatment options including adoptive T cell therapy as well as management of special situations such as chronic active EBV and EBV-associated hemophagocytic lymphohistiocytosis.
A cincuenta años de los primeros reportes de asociación del linfoma de Burkitt con el virus de Epstein-Barr (VEB), el VEB ha emergido como el tercer virus de tipo oncogénico con mayor prevalencia a escala mundial. La infección por VEB se asocia con diversas neoplasias, incluyendo el linfoma de Hodgkin y el no Hodgkin, linfoma de células T/NK y carcinoma nasofaríngeo. A pesar del control inmunológico altamente específico en el huésped inmunocompetente, el VEB puede ocasionar complicaciones severas en el huésped inmunocomprometido (es decir, la enfermedad linfoproliferativa post-trasplante). Esto es un problema particularmente en pacientes en quienes se retrasa la reconstitución de la inmunidad después de un trasplante de células madre hematopoyéticas o un trasplante de órganos sólidos. A pesar de los avances en las técnicas de diagnóstico y los algoritmos de tratamiento que permiten la identificación temprana y el tratamiento de pacientes de alto riesgo, las tasas mortalidad siguen siendo muy altas (del 90%) si no se recibe tratamiento temprano. La piedra angular del tratamiento incluye la disminución de la inmunosupresión y la depleción de células B in vivo con un anticuerpo monoclonal anti-CD20. Sin embargo, estas modalidades de tratamiento no son siempre posibles debido al rechazo del injerto, la enfermedad de injerto contra huésped y la toxicidad. Nuevas modalidades de tratamiento incluyen el uso de la terapia adoptiva de células T, que ha mostrado resultados promisorios en diversas neoplasias relacionadas con el VEB. En este artículo se revisan los avances más recientes en cuanto a los factores de riesgo, diagnóstico y tratamiento de las neoplasias asociadas con VEB, particularmente la enfermedad linfoproliferativa post-trasplante. También se discuten los tratamientos más recientes e innovadores, que incluyen la terapia adoptiva de células T así como el manejo de situaciones especiales, como la infección crónica activa de VEB y la linfohistiocitosis hemafagocítica asociada con VEB.
Epstein Barr Virus (EBV) is a highly immunogenic γ-herpes virus with a >90% worldwide seroconversion rate by young adulthood.1,2 Whereas infections in childhood are usually asymptomatic, in adolescence and early adulthood, EBV infection can manifest as acute mononucleosis, a typically self-limiting infection. During a primary infection, the normal host mounts a vigorous cellular immune response consisting of both CD4+ and CD8+ cytotoxic T lymphocytes (CTLs). These CTLs effectively control both primary EBV infection and periodic reactivations by targeting both lytic and latent cycle antigens.3 Despite the highly specific immunologic control in the immunocompetent host, EBV can cause severe complications in the immunocompromised host, particularly patients with delayed immune reconstitution post-hematopoietic stem cell transplant (HSCT) or solid organ transplant (SOT). In addition to being the primary virus associated with post-transplant lymphoproliferative disease (PTLD), endemic Burkitt's lymphoma, and up to 40% of Hodgkin (HL) and non-Hodgkin lymphoma (NHL), uncontrolled EBV infection is the cause of many HIV- or AIDS-associated lymphomas.1,4 Whereas the causative relationship between EBV and the aforementioned disorders is well established, more recently EBV viremia has been linked to hemophagocytic lymphohistiocytosis (HLH) with associated chronic active EBV infection (CAEBV).5 The common denominator in all of these scenarios appears to be the lack of EBV-specific T cells able to successfully control the infection. Whether this is due to pre-transplant conditioning regimens, the prolonged immunosuppression necessary following transplant, or anergic T cells incapable of recognizing and controlling EBV infection, all of these patients possess the perfect immunosuppressed environment for unchecked EBV reactivation and its sequelae.6
In this article we will review recent advances in risk factors, diagnosis and management of EBV-associated malignancies, particularly PTLD. We will also discuss new and innovative treatment options including adoptive T cell therapy as well as management of special situations such as CAEBV and EBV-associated HLH.
2Post-transplant lymphoproliferative disease: pathogenesis and risk factorsPTLD is a heterogeneous group of malignant diseases ranging from the classic polyclonal subtype to more aggressive, monoclonal forms. Nearly 85% of cases are of B-cell lineage, with the remaining 15% of cases of T or NK cell lineage. The majority of PTLD cases are associated with EBV infection, whereas only ∼30% of reported cases are EBV negative.7,8 EBV-negative PTLD tends to occur later in life and be monomorphic in origin (T- or NK-cell neoplasms), although the etiology of the vast majority of EBV-negative PTLD remains unknown.9
Patients are at highest risk for developing PTLD within the first year following transplant, with>80% of cases occurring within this time frame.10,11 Several characteristics make post-HSCT recipients more susceptible to PTLD. Established risk factors include transplantation from an unrelated or mismatched donor (including haploidentical or cord blood), donor-recipient serological mismatch in relation to EBV, graft T-cell depletion, use of antithymocyte globulin (ATG) and prolonged/intense immunosuppression for prevention/treatment of graft vs. host disease (GVHD).10–14 Other studies have also identified use of reduced conditioning regimens and acute GVHD ≥grade 2 as risk factors.6,15 Although the incidence of PTLD after HSCT varies in the literature, it can increase from ∼2% up to 10–20% in patients with the aforementioned risk factors.6,10,11,15
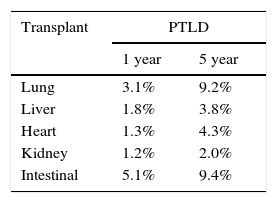
In contrast to the post-HSCT setting, the overall incidence of PTLD post-SOT has declined likely due to enhanced post-transplant quantitative monitoring of EBV viral load and subsequent adjustment of immunosuppression when indicated. Recent data from the Organ Procurement and Transplant Network (OPTN) reports a 5-year cumulative incidence of PTLD in pediatric SOT recipients of 2-9%. The highest incidence of PTLD is typically seen in lung and intestinal transplant recipients, with historical single center studies reporting an incidence in intestinal transplant recipients as high as 30% (Table 1).16 This is a reflection of immunosuppression intensity as well as the transmission of lymphoid tissue in the allograft (a potential source for primary EBV infection). Age of the transplant recipient and EBV donor/recipient mismatch are additional major risk factors. A large longitudinal study of>3000 pediatric heart transplant recipients found that 25% of EBV seronegative recipients (aged 4-7 years) receiving organs from EBV+ donors developed some form of PTLD.17 The use of lymphocyte-depleting agents and elevated tacrolimus levels has similarly been implicated in the development of PTLD.18,19
Type of transplant and risk of PTLD.
| Transplant | PTLD | |
|---|---|---|
| 1 year | 5 year | |
| Lung | 3.1% | 9.2% |
| Liver | 1.8% | 3.8% |
| Heart | 1.3% | 4.3% |
| Kidney | 1.2% | 2.0% |
| Intestinal | 5.1% | 9.4% |
Note: Cumulative 1 year and 5 year incidence of post-transplant lymphoproliferative disease (PTLD) in pediatric SOT recipients stratified by organ as reported in the 2012 OPTN/SRTR Annual Report (*Data reported combined for adults and pediatric recipients. Adapted from 2012 Annual report of US Organ Procurement and Transplantation) (Ref. 19).
Although the incidence of EBV-associated PTLD has not changed in recent years, the mortality rate can be as high as 90% if not treated early.20Table 2
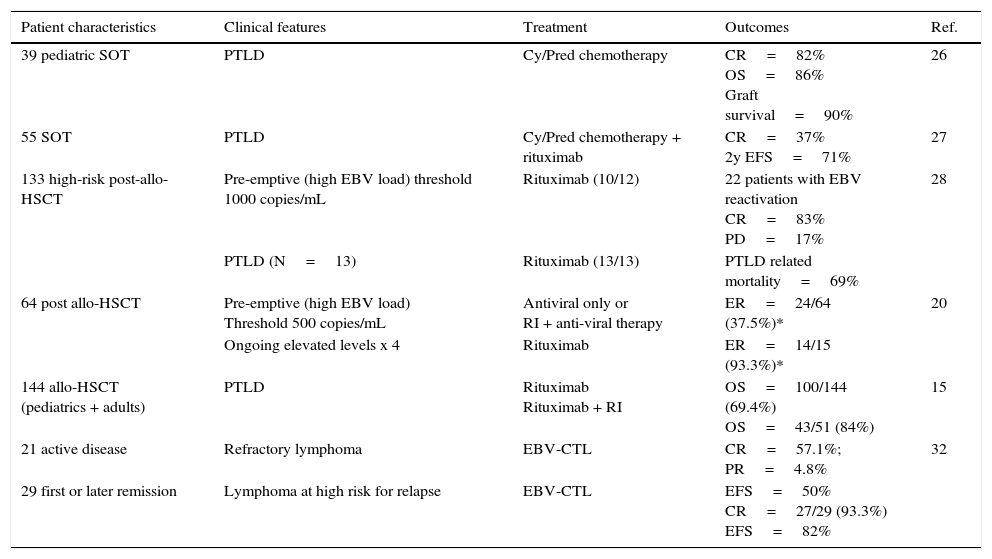
PTLD treatment and clinical outcomes.
| Patient characteristics | Clinical features | Treatment | Outcomes | Ref. |
|---|---|---|---|---|
| 39 pediatric SOT | PTLD | Cy/Pred chemotherapy | CR=82% OS=86% Graft survival=90% | 26 |
| 55 SOT | PTLD | Cy/Pred chemotherapy + rituximab | CR=37% 2y EFS=71% | 27 |
| 133 high-risk post-allo-HSCT | Pre-emptive (high EBV load) threshold 1000 copies/mL | Rituximab (10/12) | 22 patients with EBV reactivation CR=83% PD=17% | 28 |
| PTLD (N=13) | Rituximab (13/13) | PTLD related mortality=69% | ||
| 64 post allo-HSCT | Pre-emptive (high EBV load) Threshold 500 copies/mL | Antiviral only or RI + anti-viral therapy | ER=24/64 (37.5%)* | 20 |
| Ongoing elevated levels x 4 | Rituximab | ER=14/15 (93.3%)* | ||
| 144 allo-HSCT (pediatrics + adults) | PTLD | Rituximab Rituximab + RI | OS=100/144 (69.4%) OS=43/51 (84%) | 15 |
| 21 active disease | Refractory lymphoma | EBV-CTL | CR=57.1%; PR=4.8% | 32 |
| 29 first or later remission | Lymphoma at high risk for relapse | EBV-CTL | EFS=50% CR=27/29 (93.3%) EFS=82% |
Cy, cyclophosphamide; Pred, prednisone; RI, reduction of immunosuppression; CR, complete response; OS, overall survival; EFS, event-free survival; PD, progressive disease; ER, effective rate (*only those who achieved complete remission of PTLD survived); Pts, patients.
In an immunocompetent host, primary EBV infection is either asymptomatic or associated with fever, fatigue and lymphadenopathy. This initial infection is typically followed by an EBV-specific CTL-mediated response, leading to tightly controlled regulation of viral reactivation.3
In contrast, primary EBV infection or reactivation in the immunocompromised host (particularly those post-HSCT or SOT) may present as a life-threatening disease characterized by fever, lymphadenopathy, mononucleosis-like syndrome, central nervous system (CNS) disease/myelitis, pneumonia, sepsis-like syndrome and PTLD (typically associated with EBV viremia as measured by PCR).14,20 Additionally, in SOT recipients, PTLD may present as allograft failure without other symptoms.21
4Diagnosis and importance of frequent screening in at-risk patientsOne of the most challenging management questions to answer in patients with EBV-related malignancies is when to initiate treatment. In the case of rapidly progressive monoclonal variants such as diffuse large B-cell lymphoma (DLBCL)/Burkitt's or NK-T lymphoma, this question is less relevant as the clinical picture typically dictates immediate treatment. However, in patients with EBV-PTLD (whether after HSCT or SOT), the answer is less clear, making the importance of accurate and frequent screening techniques vitally important. Most institutions have the ability to measure EBV DNA level by quantitative methods. Even though the threshold beyond which EBV “DNA-emia” is associated with disease varies in the literature (with several groups suggesting a threshold of>4000 copies/μg,14 the European Conference in Infections in Leukemia (ECIL 4th) recommends weekly quantitative monitoring of EBV DNA in high-risk patients for at least 3 months following transplant. However, only 50% of post-HSCT patients with an EBV DNA level>4000 copies/μg subsequently develop PTLD.13,22 To this end, algorithms have been developed that take into account both EBV DNA load and additional risk factors to identify high-risk patients in whom the benefit of early therapy may outweigh the risks involved. In fact, Liu et al. developed a monitoring and preemptive therapy approach for EBV viremia based on duration and trend in viral load.20 Of interest, in addition to viral load and established risk factors predicting progression of EBV viremia to full-blown PTLD, the time from EBV DNA-emia to EBV-associated disease was very short (range 0-17 days, median 7 days). In our experience, the rate of rise and clinical symptomatology may indicate even more frequent monitoring is necessary. Therefore, despite the ECIL recommendation for weekly monitoring of EBV load in high-risk patients, more frequent monitoring may be necessary to allow preemptive therapy of patients at earlier stages.20 This strategy has proved valuable in the post-SOT setting as well. In a recent large survey of 71 SOT centers in Europe,>80% reported utilizing EBV DNA-emia monitoring as a means of dictating when to initiate reduction in immunosuppression. Over half of the centers queried utilized reduction of immunosuppression or a switch to mammalian target of rapamycin (mTOR) inhibitors as a therapeutic strategy23. Despite the frequency of these practices, evidence is lacking with regard to thresholds of EBV DNA-emia at which immunosuppression should be adjusted. Furthermore, inter-laboratory variation in assays for monitoring of EBV DNA-emia make a standardized approach challenging. Despite these drawbacks, the importance of monitoring the rate of EBV load rise is key to effective identification of patients at highest risk, regardless of the method.
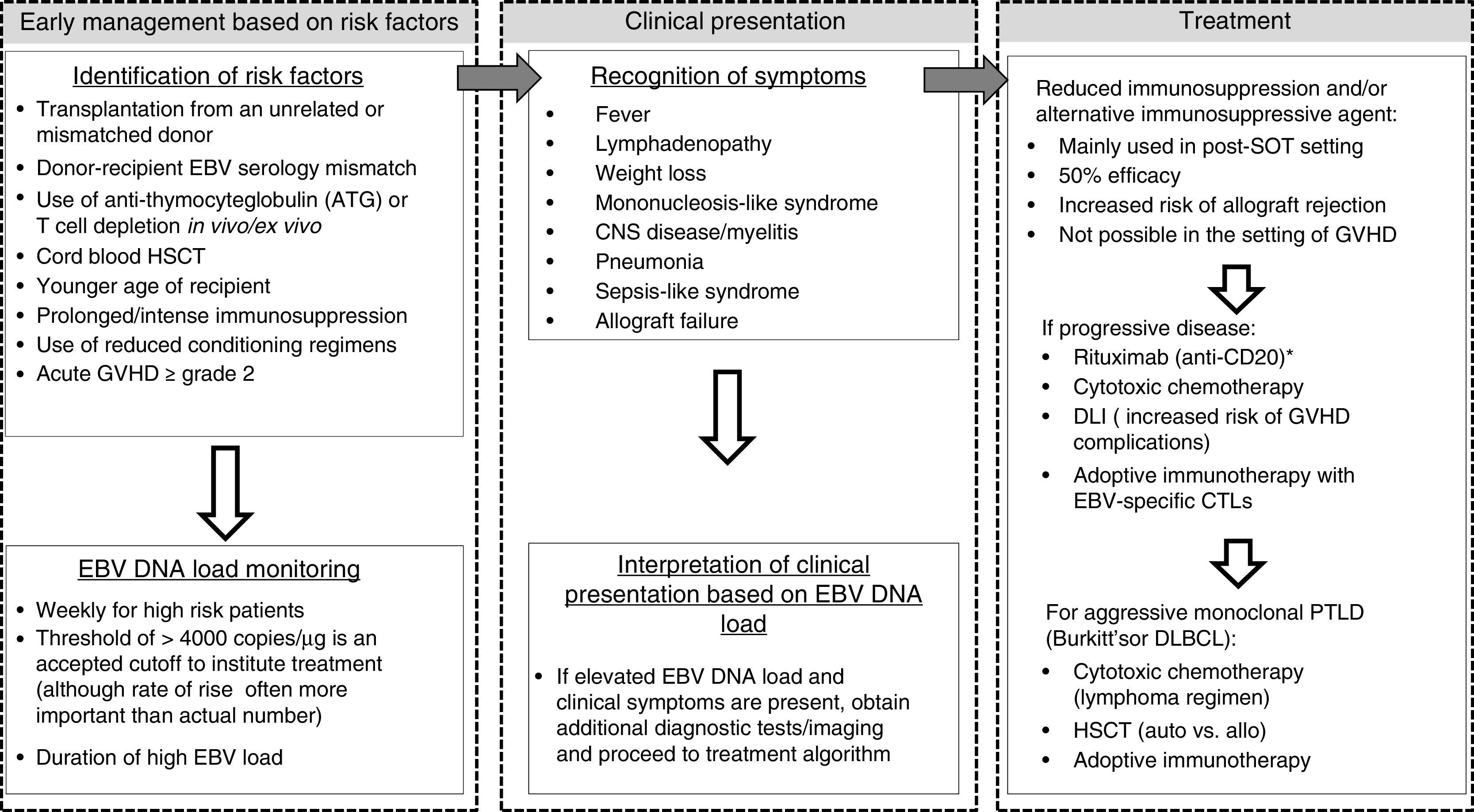
5TreatmentDespite identification of patients at increased risk for EBV viremia leading to lymphoproliferative disease, determination of how to initiate preemptive therapy remains challenging.14 Although reduction of immunosuppression alone is an effective way to re-constitute EBV-specific cellular immunity and treat PTLD (with reported efficacy rates up to 50% in some studies),22,24 particularly in the case of SOT, it carries the risk of allograft rejection, with up to half of post-heart transplant patients with PTLD treated with immunosuppression withdrawal developing acute or chronic rejection within 6 months25. When reduction of immunosuppression (RI) is not possible, other options include i) targeting pathogenic B cells using a monoclonal antibody (Anti-CD20 such as rituximab, which can yield up to 69% overall response rates),15 sometimes in combination with other cytotoxic chemotherapy, and ii) restoration of the immune response to EBV using adoptive immunotherapy. For a flow chart of the clinical management of post-transplant EBV reactivation/PTLD see Figure 1.

Flow chart of the clinical management of post-transplant EBV reactivation/PTLD. See text for details. *Rituximab may also be recommended before clinical manifestations of PTLD as pre-emptive therapy. HSCT, hematopoietic stem cell transplant; SOT, solid organ transplant; DLI, unmanipulated donor lymphocyte infusion; GVHD, graft vs. host disease; CNS, central nervous system; CTL, cytotoxic T lymphocyte; PTLD, post-transplant lymphoproliferative disease; DLBCL, diffuse large B-cell lymphoma; auto, autologous; allo, allogeneic.
Although there is no consensus regarding the optimal management of PTLD, several large studies have demonstrated that the addition of cytotoxic chemotherapy to RI +/− rituximab can be beneficial. Most chemotherapy regimens utilized for EBV-PTLD include some combination of cyclophosphamide (Cy), prednisone (Pred) and intermittent doses of rituximab. Gross et al. reported the outcomes of 39 pediatric SOT recipients who, after failing RI, received the combination of Cy (600mg/m2) and Pred (2mg/kg/day) given every 3 weeks x 6 cycles, with a complete response (CR) rate of 82%, graft survival of 90%, and overall survival (OS) of 86%26. In a larger Phase II trial of the Children's Oncology Group, 55 patients with EBV+, CD20+PTLD post-SOT (who had previously undergone a trial of RI for at least 1 week) received two initial cycles of Cy/Pred (at identical doses as the previously mentioned study) and rituximab (375mg/m2) followed by four additional cycles of Cy/Pred. Although this study reported a lower CR rate of 37%, 2-year event free survival (EFS) was 71%, indicating the potential for augmented efficacy compared to RI alone27.
5.1B-cell depletion with anti-CD20 monoclonal antibodyEven though improving the patient's immune response (by reducing immunosuppression) is one of the cornerstones of PTLD management, it may not be the best option for patients with active GVHD. Thus, eliminating B lymphocytes with a monoclonal antibody against CD20 is a feasible option. Garcia et al. evaluated the response to preemptive rituximab in 133 high-risk post-allo-HSCT recipients between the years 2006 and 2013. The study included patients receiving varying conditioning regimens [myeloablative, reduced intensity or total body irradiation (TBI) based], with similar graft manipulation and GVHD prophylaxis and at least one risk factor: HLA disparity, cord blood (CB) transplant, or use of ATG or alemtuzumab during the conditioning regimen. High-risk patients were monitored with weekly EBV qPCR from time of HSCT. Standard-risk patients were monitored weekly following the addition of a second immunosuppressive drug. The threshold for treatment with weekly rituximab at a dose of 375mg/m2 was two consecutive viral loads of>1,000 copies/mL or a single load of>2000 copies/mL. Rituximab was given until viral clearance, and then patients received an additional dose of rituximab after the virus was cleared. If there was suspicion for PTLD, a CT scan and a lymph node biopsy were obtained and if PTLD was confirmed the patient received two doses of rituximab following viral clearance.28 In this study, 16/22 patients with clinically symptomatic and histologically confirmed EBV-PTLD (ten of whom received rituximab) achieved CR for a response rate of 83%. Of note, these patients also received at least a 20% dose reduction in immunosuppression.28
Alternatively, Liu et al. created a preemptive intervention protocol based on duration and trend in EBV viral load. After detection of EBV DNA-emia in two consecutive samples (defined as ≥500 copies/ml in plasma) RI (if possible) was instituted, as well as initiation of antiviral therapy [such as ganciclovir (10mg/kg/day) or foscarnet (100mg/kg/day)]. If ongoing monitoring showed rising titers (elevated on at least four occasions), rituximab was begun. Of 251 post-allo-HSCT patients, 64 were included in the first-phase preemptive study, with 24 (37%) achieving a CR [in this study, CR was defined as a negative EBV-DNA load, or<500 copies/ml in plasma (which was the threshold for the assay used), and the absence of signs and symptoms of EBV-associated disease] and 40 with no response. Twenty five of the patients who did not respond progressed to EBV-associated disease (using the ECIL definition for clinical EBV infection). Of the 15 patients who received rituximab 14 (93.3%) had a CR. These findings suggest that although RI plus antiviral agents may be a reasonable management approach for low-risk patients, preemptive rituximab should be considered for high-risk patients. It is worth noting that although antiviral drugs may inhibit virus replication, antivirals alone (without combination with RI or rituximab) have not been shown to prevent EBV-PTLD. For this reason, the 4th ECIL does not recommend the sole use of antiviral drugs as prevention of PTLD.13,20
In another large multicenter, retrospective analysis of 4,466 allo-HSCT recipients at 19 European Transplantation centers, 144 patients were diagnosed with PTLD. Patients either received rituximab (375mg/m2 every 6-10 days; 64%) or a combination of rituximab (375mg/m2 every 6-10 days) and RI (35%); 21% of the patients required adjuvant chemotherapy due to only partial response (PR) to either rituximab alone or rituximab with additional RI. OS after rituximab alone was 69.4%; 84% of patients who received both rituximab and RI had resolution of PTLD, whereas patients who did not have RI had only 40% OS.15
Despite the effectiveness of these therapies, they are limited by toxicity and do not address the underlying deficiency in EBV-specific T cell immunity.
5.2Adoptive immunotherapyAs discussed above, the immune system controls EBV infection through CD4+ and CD8+ CTLs. EBV+ neoplastic cells express immunogenic antigens that are potential targets for CTL-mediated EBV-specific cytotoxicity. However, in the setting of significant immunosuppression and delayed immune reconstitution post-transplant, this control is inadequate.
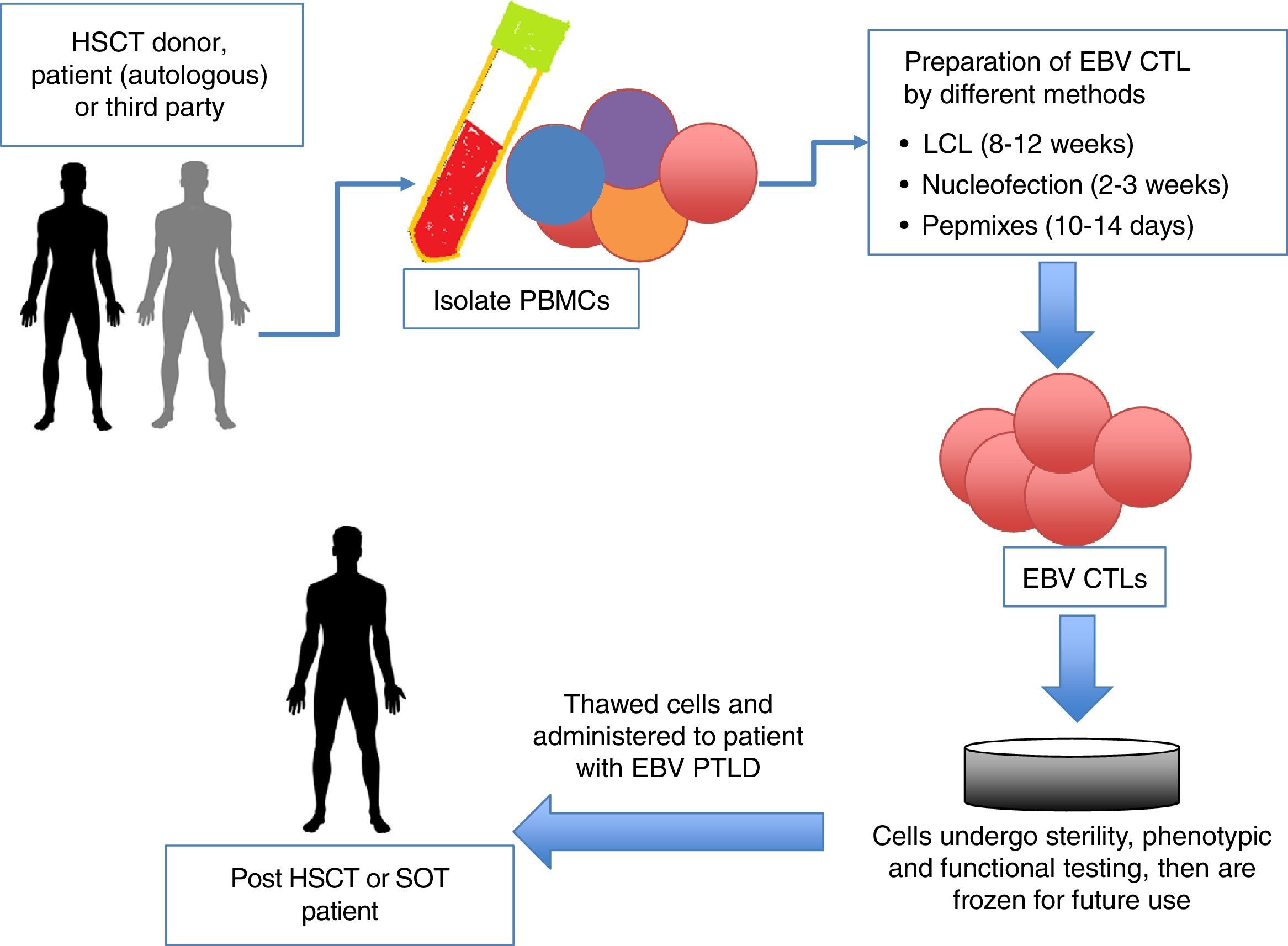
Adoptive immunotherapy with unmanipulated donor T cells and EBV-CTLs has provided well-tolerated, effective, and long-term antiviral protection.14 In the post-HSCT setting, unmanipulated donor lymphocyte infusions (DLIs) can reconstitute EBV-specific immunity with clinical response rates from 60 to 90%.29 However, GVHD is a well-known complication of DLI. Furthermore, only a minority of patients with established disease achieves sustained CRs.30 A novel and increasingly utilized approach to the treatment of EBV-PTLD is to restore the impaired immune function by the adoptive transfer of EBV-specific CTLs (Figure 2). In fact, when compared to patients receiving unmanipulated DLI, patients receiving either HLA compatible or partially HLA-matched EBV-CTLs had similar response rates (73% vs. 68% respectively).31 Because this therapy is specific for EBV-infected cells, risk of GVHD is minimal (0% vs. 17% respectively in a recent study by Doubrovina et al.31). Bollard et al. treated 50 patients with relapsed, refractory (n=21) or high-risk (due to history of multiply-relapsed disease, although in a state of remission at time of treatment) (n=29) EBV-associated HL or NHL with autologous EBV-CTLs. Of the 29 patients at high-risk for relapse (where CTLs were used as adjuvant therapy), 82% had EFS following EBV-CTL infusion, whereas 11/21 patients treated with active disease achieved CR as well. There were two PRs, with one patient achieving a CR after an additional CTL infusion.32 This approach has been employed in both the autologous (EBV-CTLs generated from the patient themselves) and allogeneic settings (cells generated from HSCT donor or, as discussed below, healthy third-party donors).29
. HSCT, hematopoietic stem cell transplant; auto, autologous; allo, allogeneic; PBMC, peripheral blood mononuclear cells; EBV, Epstein-Barr virus; LCL, lymphoblastoid cell lines; SOT, solid organ transplant; PTLD, post-transplant lymphoproliferative disease.")
Schematic diagram of adoptive immunotherapy with cytotoxic T lymphocytes (CTLs). HSCT, hematopoietic stem cell transplant; auto, autologous; allo, allogeneic; PBMC, peripheral blood mononuclear cells; EBV, Epstein-Barr virus; LCL, lymphoblastoid cell lines; SOT, solid organ transplant; PTLD, post-transplant lymphoproliferative disease.
The complexity and time taken to generate either autologous or allogeneic EBV-CTLs for adoptive transfer has been a limitation to widespread clinical applicability (manufacture time using earlier methods can take up to 12 weeks). Therefore, several groups have successfully shortened the manufacture of EBV-CTLs by eliminating the use of lymphoblastoid cell lines (LCLs) as stimulating antigen, without compromising efficacy.14 Generation methods include i) using nucleofection to transfer DNA plasmid into dendritic cells and using these as antigen presenting cells (APCs), a process that took 2-3 weeks, and reproducibly created EBV-CTLs specific for EBV antigens EBNA1, BZLF1 and LMP2 confirmed by IFN-γ ELISpot assay,29 ii) IFN-γ capture, in which the investigators selectively captured and infused the CTLs secreting the most IFN-γ in response to antigen stimulation.33 Using either of these manufacture techniques yielded promising results, with 8/10 patients achieving virological and clinical responses in the study by Gerdemann et al. although only three responses were sustained.34,35
Members of our group have successfully optimized an accelerated manufacture process using overlapping peptide libraries that allows production of virus-specific T cells (VSTs) in as little as 10-14 days. Peripheral blood mononuclear cells (PBMCs) are stimulated with overlapping peptide libraries (pepmixes) incorporating the antigens of interest, then expanded in a closed-system for 10-14 days. This manufacture method has been quite successful, with Papadopoulou et al. generating CTLs specific for five clinically problematic viruses in the post-HSCT period (including EBV) from HSCT donors; 94% of patients treated had virological and clinical responses, including patients with EBV-PTLD and reactivation, all of who achieved a CR.36
Despite the success of adoptively transferred EBV-CTLs, several groups have reported trends associated with poor clinical response.31,32 For one, failure of the EBV-CTLs to expand in vivo is associated with poor response. In the case of EBV-CTLs generated from the HSCT donor, treatment failures correlated with impaired recognition of tumor targets by the infused CTLs, mainly due to selective HLA restriction by alleles not shared by the EBV-PTLD. In fact, the Memorial Sloan Kettering (MSK) group saw encouraging clinical responses in patients who had previously failed donor-derived CTLs after choosing an alternate third-party donor with confirmed EBV-CTL activity through a shared HLA allele.31
However, despite faster manufacture time, the lack of immediate availability of EBV-CTLs highlights the need for an immediately available “off-the-shelf product”.36 This strategy is also helpful in situations where there is not a readily available donor to generate EBV-CTLs from cord blood (CB)/Matched Unrelated Donor (MUD) HSCT or post-SOT). This approach has been an active source of investigation in several centers including ours, as we work to optimize third-party partially matched VST banks for treatment of EBV-related malignancies and other viral reactivations as well. In a multicenter study, Leen et al. created a bank of third-party tri-virus T cells (active against adenovirus, cytomegalovirus and EBV) generated from healthy individuals with common HLA types, and manufactured using the LCL generation method. Cells were frozen and stored, thus available for immediate use. Fifty post-HSCT patients were infused, including eight with rituximab-refractory EBV-PTLD and one with persistent EBV DNA-emia, with a 6-week CR rate of 66.7%. Cells persisted up to 12 weeks post-infusion. Of note, clinical responses were noted even when infused CTLs were matched at only a single HLA allele, with no major GVHD reported.37
Both autologous and third-party partially HLA-matched EBV-CTLs have been used in SOT recipients as well, both as prevention and as treatment of EBV-PTLD. A single infusion of autologous EBV-CTLs in 12 pediatric heart and liver transplant recipients at high-risk for PTLD prevented development of PTLD at 1 year.38 In a study of over 30 SOT recipients with PTLD who failed conventional therapy, infusion of third party partially HLA-matched EBV-CTLs led to CR or PR in>50% of patients at 6 months.39
It is important to note that infusions of both autologous and allogeneic EBV-CTLs have been well tolerated. Specifically, there have been no reported infusion-related adverse events, significant toxicity, or graft rejection attributable to CTL infusion, and only minimal de novo GVHD. Aside from one report from our center of systemic inflammatory response syndrome (SIRS) in a patient with bulky refractory EBV lymphoma approximately 2 weeks after receiving EBV-specific CTLs, there have been no reports of cytokine release syndrome. In this patient, the inflammatory response was concurrent with in vivo expansion of the CTLs and characterized by fever, tachycardia, hypotension, respiratory distress, and elevated inflammatory markers. Symptoms resolved with steroids and etanercept.40
Although adoptive immunotherapy with EBV-CTLs is a promising approach, optimization of this therapy is dependent on having timely universal access to cellular products, not limited to specialized centers.
6Special cases6.1Chronic active EBV infection (CAEBV) and hemophagocytic lymphohistiocytosis (HLH) in the setting of PTLDIt is appropriate to discuss CAEBV and EBV-associated HLH together as the entities are thought to exist on a continuous spectrum. CAEBV, which can occur after primary EBV infection, can be of B or T cell origin. When of B cell origin, the presentation and management is similar to EBV-PTLD. When of T cell origin, it is similar in clinical features and pathologic findings to EBV-associated HLH, although EBV+HLH may progress to a monoclonal T-cell lymphoproliferative disease.5,9,41
Whereas PTLD is a complication of decreased CTL immune surveillance leading to increased susceptibility to EBV, HLH is a life-threatening condition resulting from excessive immune activation, defined by the occurrence of at least five abnormalities: fever, splenomegaly, cytopenias in at least two hematopoietic cell lineages, elevated ferritin and triglyceride levels, decreased fibrinogen or elevated soluble IL-2, impaired NK cell activity and/or hemophagocytosis on biopsy. HLH can be primary or secondary and can occur secondary to malignancy or treatment-related immunosuppression.42 Rarely, HLH occurs after HSCT (incidence 0.3%), is typically triggered by EBV, and presents with classic features of HLH. Several case reports exist detailing patients transplanted for hematologic malignancies who subsequently developed EBV-related HLH and PTLD within 100 days of transplant. Jha et al. presented a case of a 2-year-old who underwent liver transplant for extra-hepatic biliary atresia, presenting 9 months after transplant with fevers, hepatosplenomegaly, pancytopenia, EBV viremia of 934,000 copies/ml and bone marrow examination consistent with EBV-induced HLH treated with RI, steroids and rituximab achieving CR. Reported patients have been treated similarly, with rituximab, steroids, and reduction or discontinuation of immunosuppression, with symptomatic recovery after a few weeks and resolution of PTLD within months. The reported patients remain in sustained remission of their primary diseases43,44.
Although anecdotal considering the limited numbers, it appears that patients who present with concomitant PTLD and fulminant HLH post-HSCT are less likely to respond to withdrawal of immune suppression alone and will require at least the addition of rituximab or steroids.
6.2EBV-associated nasopharyngeal carcinoma (NPC)NPC is a distinctive histological subtype of head and neck cancer which is rarely seen in Western countries, but highly endemic to Southeast Asia and Southern China (incidence of 20-30/100,000) accounting for up to 20% of adult cancers in this region.45,46 Risk factors include tobacco and excessive alcohol intake. Up to 98% of NPC cases (particularly endemic) are EBV-positive.2 Treatment for early, localized disease includes radiotherapy to localized areas of disease and involved lymph nodes, with local control rates [as defined by RECIST (Response Evaluation Criteria in Solid Tumors) criteria] of 80-90%. In contrast, more advanced disease has suboptimal response to radiotherapy alone (control rate of 30-65%). However, the addition of platinum-based chemotherapy increased control rates to 54-78% in reports from the National Comprehensive Cancer Network (NCCN) and intergroup trial 0099.47
Because the majority of NPC cases express the EBV type II latency pattern (LMP-1, LMP-2 and EBNA), NPC is an ideal target for adoptive T cell therapy.48–51 Several groups, including ours, have reported promising results in the treatment of advanced NPC using EBV-specific CTL therapy. Chia et al. evaluated the safety and efficacy of chemotherapy in combination with LMP-2 specific EBV-CTLs in a Phase II clinical trial including 38 patients. After a median follow up of ∼30 months, 2- and 3-year OS rates were 62.9% and 37.1%, respectively. In fact, five patients who received CTLs did not require additional chemotherapy for>34 months following the last infusion. Treatment was well-tolerated, with no grade 3-5 toxicities, with the most common adverse effects being grade 1-2 fatigue and myalgias, transient infusion-associated fever and grade 1 skin rash.48
In a study by Comoli et al., ten patients with progressive EBV+ stage IV NPC who had failed conventional therapy received autologous EBV-specific CTLs. Patients received between two and 23 infusions, with two patients achieving PR, four patients with stable disease (lasting 4-15 months) and four with progressive disease. In three of the patients who had clinical benefit from the EBV-CTLs, increased frequencies of LMP-2 specific CTLs were detected in the peripheral blood, a phenomenon that has been noted in other studies as well.49 Louis et al. also evaluated EBV-CTLs in a Phase I/II Study of 23 patients with NPC. Seven patients were treated in the dose escalation phase of the study, and after no dose-related toxicity occurred, the remaining 16 patients were treated on the highest tolerated dose level. Of eight patients treated in remission, five remained disease free for 25-82 months. Of three patients treated with local recurrent disease, CR was achieved in two patients for>44 and>53 months, respectively. Of the 11 treated patients with metastatic disease, one achieved CR and one patient had CRu (defined as resolution of a pre-infusion imaging finding of unknown significance). The remaining patients had either PR (n=1), stable disease (n=2), or progressive disease (n=6).50
6.3Natural killer/T-cell lymphoma (NK/T)NK/T lymphomas are rare lymphomas that, in contrast to the majority of EBV-associated malignancies, typically affect the immunocompetent host. Historically, NK/T lymphoma has a very poor prognosis with 5-year survival rate of<50% with conventional chemotherapy alone.1 However, similar to EBV+ HL and NHL, the malignant cells in NK/T lymphoma express a Type II latency profile characterized by EBNA1 and LMP-2, thus making it a potential target for adoptive T cell therapy. Bollard et al. tested this approach by genetically modifying autologous T cells to increase the expression and immunogenicity of LMP-2. In this study, 9/10 patients with high-risk NKT lymphoma who received LMP-2 CTLs in a state of remission remained in remission. Strikingly, 5/6 patients with active disease had overt tumor responses, with sustained CRs (>9 months) in four patients.52 In a more recent study, 11 patients with extranodal NK/T lymphoma previously treated with chemotherapy received autologous LMP-1/2A CTLs (two cycles of four weekly doses, 1 month apart) as remission consolidation. The infusions were well tolerated, with remarkable OS and progression free survival (PFS) of 100% and 90%, respectively.53
The efficacy of LMP-CTLs as treatment of NK/T lymphoma has therefore become a viable option for a disease with historically few therapeutic options.
7When a once indolent PTLD becomes an aggressive monoclonal lymphomaUnfortunately, not all PTLD is responsive to conservative withdrawal of immune suppression, institution of less aggressive cytotoxic therapy, and adoptive immunotherapy. In some cases, a once responsive lymphoma suddenly becomes refractory, corresponding with rising levels of EBV viral load and clinical symptomatology (lymphadenopathy, fever, new or increased lesions on CT or PET scan). When medically feasible, repeat biopsy of these lesions is often necessary to determine whether a polymorphic PTLD has evolved into a more monomorphic, aggressive lymphoma such as Burkitt's or DLBCL. If biopsy confirms a more aggressive subtype, only a minority of patients will respond to RI or rituximab alone as compared to their polymorphic counterparts. For this reason, if biopsy confirms one of these more aggressive subtypes, patients will benefit from more aggressive chemotherapy-based regimens that are standard-of-care for the specific type of lymphoma. Because these cases can prove refractory to chemotherapy alone, the recommendation from the American Society for Blood and Marrow Transplant (ASBMT)54 is to refer patients who fail chemotherapy-based regimens for autologous, or in some cases, allogeneic HSCT.55
Despite the development of early-intervention-based treatment guidelines, long-term survival of patients with PTLD and other EBV-related malignancies remains suboptimal. Continued improvements in both risk stratification and identification of alternative treatment options (specifically EBV-specific CTLs) are essential to lessening the morbidity and mortality caused by EBV-associated diseases. The continued optimization of autologous EBV-CTLs and immediate availability of “off the shelf” EBV-CTLs offers the possibilityof improved access to this therapy, which will hopefully translate to improved outcomes.
Conflict of interestThe authors declare no conflict of interest of any nature.