




Este trabajo tiene el objetivo de explorar el marco normativo de las nanotecnologías (NTs) y la nanomedicina, y elaborar un análisis comparativo entre dos regímenes normativos: los Estados Unidos y la Unión Europea. En el artículo se identifican y clasifican las posturas que permiten el diseño de un marco regulatorio de las NTs. Además, se comparan dos regímenes normativos: 1) sustancias químicas, y 2) medicamentos y productos sanitarios. El objetivo principal es reconocer si la regulación actual es eficiente, suficiente y apropiada para las NTs. En el primero se contrastan tres marcos jurídicos: los Estados Unidos, la Unión Europea y la Organización Internacional de Normalización. En el segundo régimen se compara la Agencia Europea de Medicinas de la Unión Europea y la Administración de Medicamentos y Alimentos de los Estados Unidos. Se concluye que la regulación actual en materia de sustancias químicas es todavía limitada para el caso de las NTs, debido a que hay procesos que no se abordan o supuestos que no aplican para las NTs. Igualmente, para las NTs, la regulación en sustancias químicas/medicamentos y productos sanitarios puede magnificar los vacíos legales existentes. Asimismo, para el caso de la nanomedicina y las NTs, la categorización en medicamentos y aparatos médicos resulta inapropiada, ya que existen productos de combinación que pueden estar en ambas categorías.
This paper aims to explore the regulatory framework of nanotechnologies (NTs) and nanomedicine, and develop a comparative analysis between two regulatory regimes: the United States and the European Union. It identifies and classifies the positions that allow the design of a regulatory framework for NTs. As well, two regulatory regimes are compared: 1) chemicals, and 2) drugs and medical devices. The main objective is to acknowledge if the current regulation is efficient, sufficient and appropriate for NTs. In the first, three legal frameworks are compare: the United States, the European Union and the International Organization for Standardization. In the second regulatory regime the European Medicines Agency of the European Union and the Food and Drug Administration of the United States are compared. We conclude that the current regulation on chemical substances is still limited to the case of NTs, because there are processes that are not addressed or assumptions that do not apply to the NTs. In addition, in the case of NT, the separation in the regulation of chemicals and drugs and medical devices can magnify existing loopholes. Similarly, in the case of nanomedicine and NTs, categorization in drugs and medical devices might be inappropriate, since there are combination products that can be both.
Uno de los constantes debates que persevera es si es posible diseñar una regulación que busque la protección de la sociedad, sin inhibir los caminos de la investigación y el desarrollo tecnológico; esto es conocido en la literatura como “el dilema de Collingridge”.1 Las tensiones crecen aún más cuando se trata de regular procesos que todavía no están plenamente en ejecución y que se ignora y cuestiona su verdadero alcance y consecuencias; tal situación es el caso de la investigación en nanotecnologías.
El hilo conductor de este trabajo, en un sentido amplio, es la regulación de las nanotecnologías. Específicamente, se comparan los marcos normativos de Estados Unidos, la Unión Europea y la Organización Internacional de Normalización, con el objetivo de conocer si dicha regulación es eficiente y adecuada para las NTs. Es necesario mencionar que se eligen estas dos jurisdicciones, ya que es viable establecer puntos de partida y categorías de análisis. El argumento central es analizar tres diferentes visiones normativas y el marco institucional con el que funciona la regulación en nanomedicina.
Para ello se realiza, en primer lugar, una revisión de literatura respecto a las distintas posturas teóricas en relación con el diseño de un marco normativo propiamente para las NTs. Dichas posturas se identifican y clasifican en fondo y forma.
Posteriormente, se comparan dos regímenes normativos: 1) sustancias químicas, y 2) medicamentos y productos sanitarios. En el primero se contrastan tres marcos jurídicos: a) el Reglamento de Registro, Evaluación, Autorización y Restricción de Sustancias Químicas de la Unión Europea (REACH, por sus siglas en inglés), cuya facultad reside en la Agencia Europea de Sustancias y Mezclas Químicas (ECHA, por sus siglas en inglés); b) la Ley de Control de Sustancias Tóxicas de los Estados Unidos (TSCA, por sus siglas en inglés), que está bajo la autoridad de la Agencia de Protección Ambiental (EPA, por sus siglas en inglés), y c) la Organización Internacional de Normalización (ISO, por sus siglas en inglés) como marco jurídico internacional. El segundo régimen normativo a comparar es la Agencia Europea de Medicinas de la Unión Europea (EMA, por sus siglas en inglés) y la Administración de Medicamentos y Alimentos de los Estados Unidos (FDA, por sus siglas en inglés). Ambos órganos están encargados de regular los medicamentos y productos sanitarios.
Finalmente, concluimos que la regulación actual en materia de sustancias químicas es limitada para el caso de las NTs, debido a que hay procesos que no se abordan o supuestos que no aplican para las NTs. Además, para el caso de las NTs, la separación en la regulación de sustancias químicas y medicamentos y productos sanitarios puede magnificar los vacíos legales existentes. Igualmente, para el caso de la nanomedicina y las NTs, la categorización en medicamentos y aparatos médicos resulta inapropiada, ya que existen productos de combinación que pueden estar en ambas clasificaciones.
IINociones de nanotecnologíaHay distintas instituciones que han emprendido la tarea de construir una definición de “nanotecnología” (NT). Una de ellas es la Iniciativa Nacional de Nanotecnología de los Estados Unidos (NNI, por sus siglas en inglés), que define NT como “el conocimiento y el control de la materia en dimensiones entre aproximadamente 1 y 100 nanómetros (nm), donde los fenómenos únicos permiten novedosas aplicaciones (traducción propia)”.2 La Organización para la Cooperación y el Desarrollo Económico (OCDE) considera a la NT como “un conjunto de tecnologías que permiten la manipulación, estudio o explotación de pequeñas estructuras y sistemas (traducción propia)”.3
Para el caso de la Unión Europea, la Comisión considera a la NT como una “ciencia y tecnología a escala nanométrica de los átomos y moléculas, y a los principios científicos y las nuevas propiedades que pueden ser comprendidas y controladas cuando se interviene a dicha escala (traducción propia)”.4 Además de determinar que se entiende por NT, la Comisión define como nanomaterial: ...un material natural, secundario o fabricado que contenga partículas, sueltas o formando un agregado o aglomerado y en el que el 50% o más de las partículas en la granulometría numérica presente una o más dimensiones externas en el intervalo de tamaños comprendidas entre 1 nm y 100 nm. En casos específicos y cuando se justifique por preocupaciones de medio ambiente, salud, seguridad o competitividad, el umbral de la granulometría numérica del 50% puede sustituirse por un umbral comprendido entre el 1% y el 50%.5
No obstante, la definición está siendo revisada por la Comisión, con el objetivo de integrar y considerar los avances científicos y tecnológicos. El Joint Research Center (JRC) de la UE ha publicado tres reportes de la serie titulada “Towards a Review of the EC Recommendation for a Definition of the Term «Nanomaterial»”. El reporte más reciente de 2015, “Scientific-Technical Evaluation of Options to Clarify the Definition and Facilitate its Implementation”, señala que la actual definición apoyada en el tamaño como la única propiedad definitoria de una nanopartícula es la conveniente. Existe la necesidad de aclarar algunos términos utilizados en la definición para una mayor utilidad en su aplicación, como “partícula”, “dimensión externa” y “partícula constituyente”. Entre todas las cuestiones tratadas en el informe, una recomendación esencial para la implementación de la definición es el desarrollar un método para probar cuando un material no es un nanomaterial.6
Dicha escala se entiende de la siguiente manera: un nanómetro (nm) es igual una mil millonésima parte de un metro. Por ejemplo, una hoja de papel es aproximadamente 100,000 nanómetros de espesor; un cabello es 80,000 nm de ancho; una célula de cáncer es de entre 10,000 a 100,000 nm; una bacteria en promedio es de 1,000 nm; una célula de sangre es de 5 nm de diámetro; una molécula de glucosa es 1 nm.7
En particular, y de acuerdo con la Fundación Europea de la Ciencia, la nanomedicina se puede definir como el uso de la NT para mejorar la asistencia médica. Por muchos especialistas se considera que la mayor aportación de la NT estará en la nanomedicina, es decir, en el desarrollo de nuevos y efectivos tratamientos médicos.8 Puthli y colaboradores consideran a la nanomedicina, en sentido amplio, como la aplicación de las tecnologías de nanoescala a la medicina, y enfocada al desarrollo de sistemas de administración de fármacos, llamados nanofarmacéuticos.9
De esta manera, la propia definición del objeto a regular está todavía en construcción, aun cuando diversas aplicaciones en NT ya están al alcance de la sociedad y de los consumidores. Esto representa un desafío institucional y jurídico, pero especialmente constituye un espacio de opacidad en las leyes en diferentes países, lo que dificulta, incluso, el debate mismo sobre regulación.
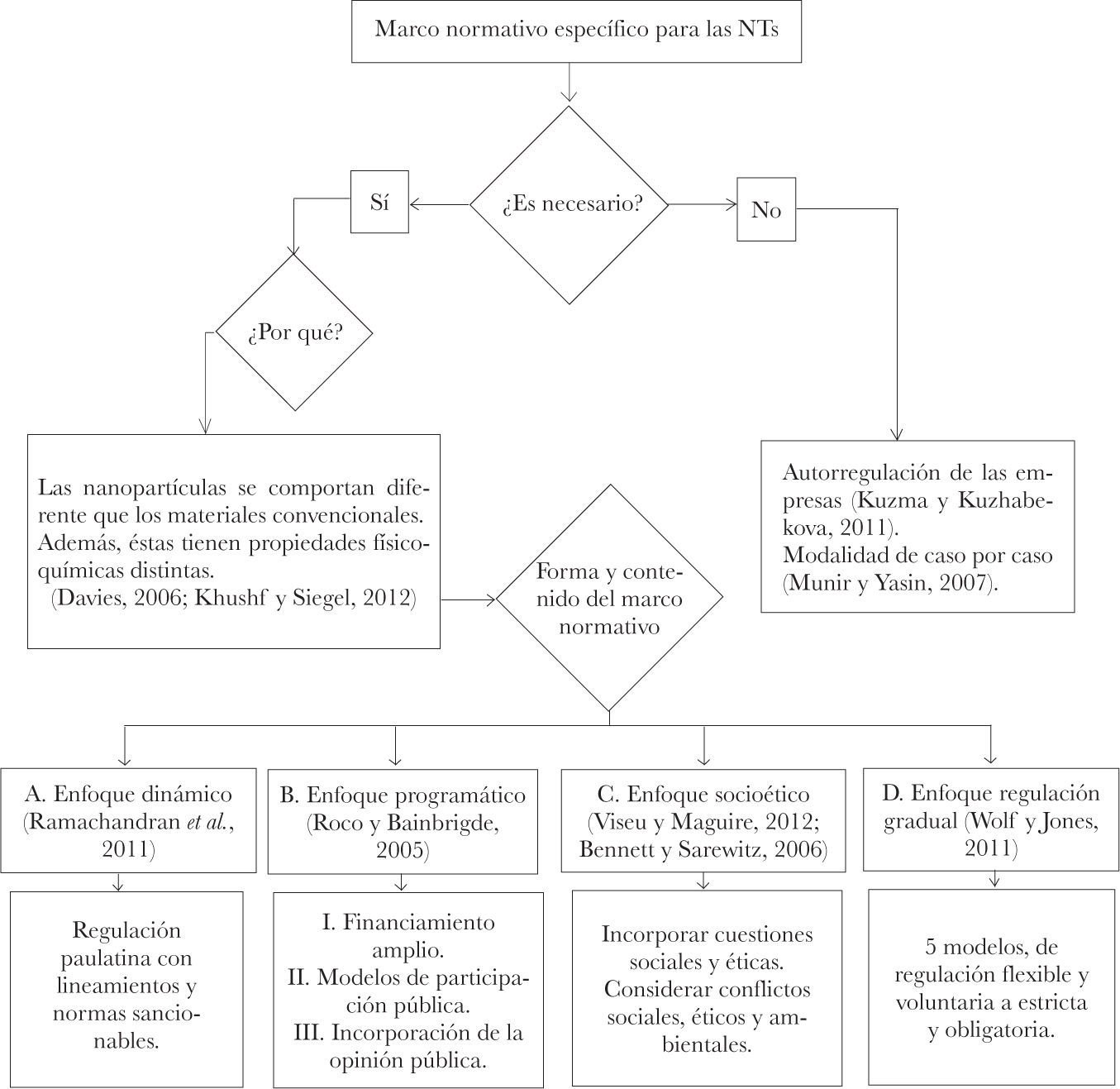
IIIRevisión de literatura en relación con las perspectivas teóricasEn la discusión respecto a la regulación podemos identificar diferentes enfoques en un debate todavía abierto y muy dinámico. Un primer grupo de ideas gira en torno a la pregunta de si es necesario una regulación específica a las NTs o si es suficiente con la regulación existente. Davies argumenta que es necesario contar con una categoría de regulación propia a las NTs. Esto se debe a que (1) los nanomateriales se comportan diferente que los materiales convencionales, y (2) a que las propiedades de los nanomateriales a menudo no son predecibles a partir de las leyes de la química y la física clásica.10 En este sentido y debido a la posibilidad de que una nanopartícula forme parte de un sistema biológico y se reevalúen las posibilidades de tratamientos e intervenciones médicas, es necesario diseñar un marco legal propio.11
Asimismo, Davies señala que “el futuro de la población mundial estará determinado por las nuevas tecnologías, pero regularmente no existe una oportunidad para que las personas consideren qué tecnologías deberían ser promovidas o desechadas y cómo hacer frente a las consecuencias e impactos de una tecnología en particular (traducción propia)”.12 Por ello, es necesario desentrañar las potencialidades positivas y negativas de las nuevas tecnologías y aplicar la ética de una forma dinámica, reevaluando continuamente las circunstancias.13 Debido a la propia naturaleza y evolución de la tecnología y de la sociedad, es necesario alinear el marco jurídico a esta dinámica para así tener una regulación lo más eficiente posible. Sin embargo, Kuzma y Kuzhabekova consideran una perspectiva teórica contraria a la antes señalada, donde se tome en cuenta la “...responsabili-dad social de las empresas como un aspecto importante en un sistema de regulación integral para la NT (traducción propia)”.14 Estos autores afirman que la incertidumbre y el rápido desarrollo de las nuevas tecnologías hacen difícil el diseño de un marco normativo eficaz. Por esta razón, el control sobre los potenciales riesgos (si no todos) se debería quedar en manos de las empresas.15 En esta misma línea, Swiss Re apoya este enfoque y considera que el actual marco jurídico es suficiente, ya que las leyes existentes de responsabilidad se aplican a los riesgos potenciales de las NTs. En caso de ser necesaria una regulación, debería de ser caso por caso y de carácter secundario.16
Posteriormente, se tienen las posturas teóricas, tanto académicas como institucionales, que consideran imperiosa la necesidad de un marco jurídico propio a las NTs. La discusión se centra en la forma y contenido de dicho marco. A continuación se analizan cuatro diferentes enfoques que presentan alternativas para regular las NTs.
La primera perspectiva es el “enfoque dinámico” propuesto por Ramachandran y colaboradores, quienes, con apoyo de la Fundación Nacional de Ciencia de los Estados Unidos (NSF, por sus siglas en inglés), analizaron del actual marco jurídico de este país. Ellos concluyeron que, en efecto, existe la necesidad de crear un marco legal específico para nanomedicina y nanobiotecnología; a su vez, sostienen que debido a la complejidad y diversidad en la nanomedicina, la regulación de estas tecnologías deberá desarrollarse a través del tiempo, utilizando herramientas de supervisión tanto rígidas (es decir, normas con facultad de sanción) como flexibles (entre ellas se incluyen recomendaciones o lineamientos a seguir).17
Guerra hace hincapié en la normativa flexible debido a la falta de información y conocimiento del tema; no obstante, señala que se debe contar con estándares para realizar exámenes premercado y principios que consideren cuestiones sociales y consentimiento público.18 En este mismo sentido, Marchant y Sylvester se inclinan por un marco normativo flexible a través de diversos instrumentos, como códigos de conducta, estándares de consenso internacional, diálogo transnacional, etcétera. Ellos reconocen la necesidad de un marco jurídico internacional propio de las NTs. 19
La necesidad de un marco normativo de carácter internacional es discutido por varios autores debido a la competencia regulatoria que puede presentarse entre las corporaciones en el desarrollo de nanotecnologías, aunque —como se ha demostrado en otros temas regulatorios— es difícil de alcanzar una regulación internacional armonizada, sin la necesaria y coincidente voluntad política de algunos de los más importante actores; sin embargo, señalan que de alcanzarlo, ello será en un futuro muy lejano. Como resultado de la falta de regulación de las NTs, ahora están surgiendo una serie de fisuras o vacíos legales.20
Roco y Bainbridge presentan una segunda perspectiva para la regulación de las NTs. Ellos sostienen un enfoque programático, en el cual indican que la investigación en las NTs debe contar con tres características: (1) financiamiento amplio... protocolos presentados por el investigador y revisados por pares [y] ...el financiamiento no debe ser impulsado por una serie de prioridades específicas de arriba hacia abajo, (2) apoyo para desarrollar varios modelos de participación pública e interacción, para establecer mejores prácticas para la educación, la comunicación y la participación de diversos públicos respecto a la NT, e (3) incorporar la continua participación del público en las deliberaciones sobre NT para asegurar el intercambio de las dos vías entre nanocientíficos e ingenieros y el público (traducción propia).21
De acuerdo con ellos, este enfoque ayudará en la investigación y desarrollo (I&D) de las NTs, ya que facilitará alianzas entre industria, universidades, laboratorios nacionales, organizaciones internacionales y organismos. No obstante, pueden surgir problemas en las sinergias mencionadas, debido a que la industria (conformada por compañías o grandes corporaciones) podría favorecer los códigos de conducta voluntarios en lugar de normas obligatorias, especialmente cuando se pretende no afectar el objetivo del status quo: ganancia. Ante ello, el propósito de garantizar la seguridad del producto podría no ser la prioridad, y se tomará en cuenta solamente una vez que se encuentre en el mercado.22 Este enfoque, efectivamente, puede promover y acelerar la I&D de las NTs, pero será en menoscabo de la garantía de salud y seguridad humana y ambiental.
Un tercer enfoque es presentado por Viseu y Maguire, quienes consideran el incorporar cuestiones sociales y éticas a la investigación y al desarrollo de la ciencia. Para ellos, las partes interesadas no deben minimizar los problemas; por el contrario, tienen que analizar y desentrañar las complejidades de las NTs y descubrir las relaciones entre ciencia y sociedad que involucran a los responsables de la salud pública.23 A pesar de que la ciencia, la tecnología y la sociedad (CTS) comparten un vínculo constante, la sociedad no juega un papel externo en el desarrollo de la C&T; en realidad, la tecnología expresa los valores y objetivos de la sociedad en la que se crea, y por lo mismo influye en ellos.24 Asimismo, en este tercer enfoque, Bennett y Sarewitz subrayan la importancia de considerar y discutir las inquietudes y conflictos sociales, éticos y medioambientales.25
Wolf y Jones presentan un cuarto enfoque, que identifica cinco modelos de regulación adicional. El primer modelo es el de la innovación impulsada a nivel local, donde las autoridades locales se encargan de crear normas o protocolos adicionales. El problema con este modelo es que se pueden presentar conflictos de jurisdicción cuando la investigación se extienda a otras regiones; no obstante, podría ser beneficioso contar con normas específicas, en lugar de normas ordinarias cuya generalidad descuide la regulación adecuada. El segundo modelo es una supervisión federal para un marco jurídico local. Este modelo evita generalidades al proporcionar una supervisión adicional para una área de investigación específica. El tercer modelo agrega al segundo la posibilidad de referirse a una agencia federal o paneles de revisión y aprobación. El cuarto y quinto consideran una agencia federal, cuyo fin es proporcionar orientación para la investigación. La diferencia entre estos dos radica en que el cuarto modelo presenta un órgano de supervisión federal permanente específico para ciertas áreas o tipos de investigación. Por su parte, el quinto modelo implica una revisión y aprobación federal obligatoria. Por lo tanto, el grado de supervisión aumenta conforme se avanza en los modelos, donde el primero es un estándar y el quinto tiene parámetros más exigentes.26
Además de contar con cuatro enfoques para determinar el marco jurídico apropiado para las NTs, varios académicos antes citados coinciden en buscar un equilibrio entre la innovación tecnológica y la salud humana. Ramachandran y colaboradores, así como Chau y colaboradores, aceptan que uno de los principales retos para una regulación de la nanomedicina es llegar a un equilibrio adecuado entre apoyar la innovación versus el mantenimiento de la salud y la seguridad pública.27 Ellos consideran que la regulación no debe ser demasiado rígida o demasiado flexible. Munir y Yasin coinciden en que debe haber equilibrio entre fomentar la innovación y promover un acceso oportuno del paciente a los beneficios de la nanomedicina y el garantizar la seguridad de los pacientes y los trabajadores de la salud.28 La noción de acceso oportuno del paciente es importante, porque el “retrasar el uso de tecnologías que salvan o mejoran vidas (life-saving or life-enhacing) puede ser tan perjudicial como la liberación prematura de tecnologías riesgosas... (traducción propia)”.29
En este sentido, Allhoff mantiene que “los beneficios conferidos por la aplicación de las NTs a la administración de fármacos superan los riesgos, a pesar de que éstos en la administración de fármacos son probablemente mayores que los generados por las nanocirugías (traducción propia)”.30 Incluso, si los beneficios superan los riesgos, es esencial realizar una evaluación de riesgo-beneficio o coste-eficacia. Sin embargo, la dificultad de la evaluación es uno de los problemas; no obstante, el público debe estar seguro de que cuando una aplicación —es decir, los productos de la na-nomedicina— se encuentra en el mercado, el gobierno ya ha tomado las medidas necesarias para proteger el medio ambiente y la salud humana, sin restringir u obstaculizar nuevas industrias y tecnologías.31 Se pretende alcanzar este equilibrio, en donde, por un lado, está la protección de la salud humana, el acceso y la seguridad del paciente, la salud de los trabajadores, y la salud pública, y por el otro, se encuentra la promoción de la investigación, innovación y nuevas industrias. En este sentido, cómo alcanzar un comportamiento responsable de todas las partes interesadas, pero sobre todo del gobierno, buscando compaginar las prioridades y objetivos. A este desafío hay que añadir la complejidad de la tecnología y la sociedad en general, lo que podría hacer aún más difícil predecir las consecuencias de las innovaciones a largo plazo.32
Uno de los aspectos centrales en todo este debate es entender qué valores o intereses se expresan en la investigación de la nanomedicina, es decir, precisamente su contenido ético-filosófico. Umbach cuestiona que una investigación, en tanto que se lleva a cabo con determinado financiamiento, está limitada por los objetivos de un cierto grupo de intereses o de una poderosa elite;33 sin embargo, son los gobiernos, como garante de derechos y obligaciones, quienes deben asegurar que el financiamiento para el desarrollo de la investigación no sea acaparado por una elite. A su vez, se podría y se debería buscar involucrar a todas las partes interesadas en el diseño de un marco legal. Sandler cuestiona la idoneidad “para asignar financiamiento, experiencia, personal y recursos de infraestructura en el desarrollo de tecnologías... con el potencial de aumentar o mejorar la calidad de vida cuando... hay personas que no cuentan con conocimientos y práctica en atención sanitaria... o incluso con la atención básica de salud (traducción propia)”.34
No obstante, en el contexto actual el financiamiento de la investigación se encuentra directamente vinculada a la búsqueda y obtención segura de ganancia, donde la nanomedicina puede llegar a ser una de las aplicaciones más rentables de las NTs. Sin embargo, uno de los problemas centrales es que las empresas que están en ello, es decir, generando aplicaciones en nanomedicina, las generan dejando de lado muchos de los intereses de la población, y especialmente de la población de los países en desarrollo. Un ejemplo claro es cuando se compara la cantidad de recursos financieros dedicados a los medicamentos para la disfunción eréctil en los Estados Unidos contra la ausencia de inversión en la investigación para los medicamentos contra la malaria.35 Si la ganancia es la única consideración para la determinación de las áreas de investigación y aplicaciones de la nanomedicina, esta dirección de la investigación y de la tecnología está exacerbando las desigualdades económicas y sociales, y también las disparidades entre los países desarrollados y en desarrollo y al interior de ellos mismos.
La persistente presencia de riesgos, problemas y efectos que engloba el uso de la nanomedicina hace imperativo la formulación de un marco legal. De esta manera, Allhoff considera que hay dos conflictos: 1) los riesgos de toxicidad y seguridad, y 2) la justicia distributiva.36 No obstante, Sandler señala que estos dos no son los únicos problemas de la nanomedicina, pues ésta tiene que enfrentarse a problemas éticos, sociales y legales.37 Entre los principales problemas de las NTs se encuentran las implicaciones sociales, las cuales han sido las más estudiadas en la literatura. Empero, retrasar la investigación tiene sus consecuencias; por ejemplo, podría terminar impidiendo el desarrollo de innovaciones tecnológicas que ofrezcan una oportunidad para aumentar la salud y el bienestar.38
De este modo, varias agencias e instituciones han tratado de fijar algunos puntos que resultan muy relevantes para la regulación, tratando precisamente de contestar las siguientes preguntas: cuánto y de qué forma la nanomedicina podría cambiar la vida de los seres humanos; cuánto y de qué manera es posible regular su investigación y aplicaciones para preservar sus mejores resultados, y al mismo tiempo proteger a las sociedades de sus posibles efectos negativos.
El siguiente cuadro sintetiza los distintos enfoques teóricos analizados previamente.
IVRegulaciones en materia de sustancias químicas, medicamentos y productos sanitarios
Como se expuso en el apartado anterior, uno de los debates teóricos centrales se enfoca en cuestionar si el marco normativo existente es adecuado para regular las NTs. Parece haber un consenso en la necesidad y falta de conocimiento sobre las NTs; esto significa que no se conocen a profundidad algunos elementos esenciales para su regulación, como propiedades químicas y físicas, riesgos de toxicidad, evaluación de riesgos y evaluación riesgo-beneficio en la manufactura, costo y seguridad.
No obstante, los retos que enfrenta la regulación en el caso de las NTs, incluyendo la nanomedicina, son similares a aquellos que presentan otras tecnologías emergentes; entre ellos se encuentran los métodos de medición y control, disponibilidad de expertos y lineamientos. Los Estados Unidos y la Unión Europea, en particular, han retrasado la adopción o modificación de la regulación dentro de los marcos normativos de sustancias químicas.
De acuerdo con Justo-Hanani y Tamar: Mucha de la literatura existente sobre regulación de la nanotecnología se centra en la falta de capacidad o voluntad política de los Estados; la evidencia muestra una ruta adaptada en la formulación de políticas regulatorias, en las cuales las instituciones de los Estados Unidos y la Unión Europea ejercen un poder centralizado dentro de sus políticas para identificar e influenciar las trayectorias tecnológicas para conseguir los fines socialmente deseables.39
En este trabajo se analizan tres marcos jurídicos: el europeo, a través de la Agencia Europea de Sustancias y Mezclas Químicas (ECHA, por sus siglas en inglés); los Estados Unidos, por medio de la Agencia de Protección Ambiental de los Estados Unidos (EPA, por sus siglas en inglés), y también el marco normativo internacional, mediante la Organización Internacional de Normalización (ISO, por sus siglas en inglés).
En lo que respecta a la regulación de sustancias químicas, en la Unión Europea el REACH se encarga de valorar los impactos negativos de éstas, en este caso de nanopartículas, que puedan afectar la salud humana y el medio ambiente. Sin embargo, la regulación actual presenta vacíos legales en materia de NTs, puesto que —por ejemplo— en el caso de la exportación e importación de sustancias químicas, ésta regula solamente en los casos en los que la producción sea mayor a cien toneladas. El problema surge cuando la producción de nanomateriales está por debajo de cien toneladas.40 Es necesario resaltar este vacío legal, ya que los nanomateriales casi no se comercializan por toneladas, lo que resulta en sustancias químicas comercializadas no reguladas.
El problema de armonización entre las regulaciones que pertencen a diferentes campos reitera la necesidad de una normativa propia.41 Por un lado, ECHA y EPA en la Unión Europea y los Estados Unidos, respectivamente, regulan las sustancias químicas, dentro de las cuales se encuentran algunas nanopartículas.
Regulación de sustancias químicas4242 Elaboración propia a partir de Justo-Hanani, R. y Tamar, D., op. cit.; Gispert, I., “Overview of Nanomedicines Regulation in the European Union”, Frontiers of Nanoscience, vol. 4 (1), 2012, pp. 487-507; ISO, “International Organization for Standardization”, 2014.
| Unión Europea | Estados Unidos de América | Internacional |
|---|---|---|
| reach | epa | iso |
| Órgano o regulación | ||
| Registration, Evaluation, Authorization and Restriction of Chemicals (EU) (European Chemical Agency, EChA). | Environmental Protection Agency (US) (Toxic Substance Control Act-TSCA). | International Organization for Standarization. |
| Enfoque | ||
| Regulación compleja para proteger la salud humana y medio ambiente: mejorar y promover la competitividad y libre circulación de sustancias. Autoridad Central (CE) delega de facultades (EChA). | Voluntario, a discreción de los fabricantes. Aunque la EPA intenta constantemente limitar esta discreción y aumentar su autonomía normativa. | Estandarización de normas de productos y seguridad para las empresas u organizaciones a nivel internacional. |
| Antecedentes/historia | ||
| En vigor desde 2007. El proceso de registro se llevará a cabo en tres etapas a lo largo de once años. | TSCA IUR rule, en vigor desde 1986. | El ISO que trata temas en relación con nanomedicina es el 80004-5 y 7. |
| 2009. El Parlamento implementó el principio “no hay datos no hay mer-cado” (“no data no market”) a través de un sistema de información obligatorio para los fabricantes. | Regla TSCA IUR otorga la facultad a EPA para requerir informes a la industria cada cinco años. | Ambos son especificaciones técnicas. |
| 2010. EChA (European Chemical Agency) ingresó a un inventario de nano (“nano inventory”) que complementa REACH. | Enero de 2008. The EPA's Nanoscale Material Stewardship Program (NMSP), plan de información voluntaria respaldado por el compromiso global de la industria. | A) ISO/TS 80004-5:2011 Nanotechnologies —Vocabulary— Part 5: Nano/bio interface. Establece términos y definiciones con respecto a la interrelación entre nanomateriales y biología. Destinado a facilitar la comunicación entre científicos, ingenieros, fabricantes, reguladores, ONG y consumidores. |
| 2011. La Comisión llegó a un acuerdo acerca de una definición única para garantizar la conformidad a través de áreas legislativas y sectores en el comercio de nanomateriales. | 2010. Se aprobaron estrictos requisitos de divulgación para nanoestructuras de carbono, principalmente, con uso comercial (Multi-Walled Carbon Nanotubes and Single-Walled Carbon Nanotubes-CNTs). | B) ISO/TS 80004-7:2011 Nanotechnologies —Vocabulary— Part 7: Diagnostics and therapeutics for healthcare, Nanotechnologies. Aplica el uso de nanotecnologías en el diagnóstico médico y terapia. Proporciona términos claros y constantes para profesionales de la salud, fabricantes, consumidores, técnicos, agentes de patentes, reguladores, ONG e investigadores. |
| 2014. La Comisión acuerda en dos métodos para determinar propiedades físico-químicas, evaluación de sustancias persistentes, bioacumulativas y tóxicas, evaluación de actividad endócrina, toxicocinética, prueba de toxicidad crónica y carcinogenicidad combinado. | 2012. La regulación se extiende a nanoestructuras de uso genérico que están sujetas a un proceso de revisión de prefabricación (PMN: premanufacture review process) o también referidas como sustancias químicas de interés. | |
| Diferencias | ||
| Las decisiones del Parlamento sirven como modelos para las iniciativas de ley de la Comisión. | Carece de apoyo en el Congreso y de liderazgo en la formación de normatividad o principios normativos para el mercado de los nanomateriales a nivel mundial. | Organización internacional que establece estándares voluntarios. |
| Estándar internacional de facto para la creación de mercados. Dichos estándares regulan el acceso al mercado para el mercado mundial en su conjunto. | Se enfoca en normas de información de datos nacionales. | |
| Las instituciones de la Unión Europea han utilizado una autoridad más formal para reconfigurar el papel de los actores del mercado mundial. | El mandato de la autoridad federal de Estados Unidos bajo TSCA es limitado. | |
| La Comisión rechazó una normatividad privada. | El enfoque de EPA sobre las políticas de nano se limita a mejorar el cumplimiento normativo, sin restricciones de creación de mercados o autoridad de actores privados. | |
| Investiga los siguientes materiales: nanotubos de carbono, óxido de cerio, dióxido de titanio, nanoplata, hierro, cobre micronizado. | ||
Por otro lado, la Administración de Alimentos y Medicamentos y la Agencia Europea de Medicinas (EMA) regulan los aspectos médicos que incluyen NTs. Estos marcos jurídicos se encargan de normar los medicamentos y los productos sanitarios. En el siguiente cuadro se muestra un estudio comparativo de dos marcos normativos en materia de medicamentos y dispositivos médicos: el de la Unión Europea y el de los Estados Unidos.
Regulación de medicamentos y productos sanitarios4343 Elaboración propia a partir de Anatol, R. et al., “Continuing to Strengthen FDA's Science Approach to Emerging Technologies”, Nanomedicine: Nanotechnology, Biology and Medicine, 9 (5), 2013, pp. 594-599; Directive 90/385/EEC; Directive 93/42/EEC; Directive 98/8/EC; Directive 98/79/EC; Directive 2002/98/EC; Directive 2004/23/EC; Directive 2004/27/EC; Directive 2007/47/EC; D'Silva, J. y Van Calster, G., “Taking Temperature. A Review of European Union Regulation in Nanomedicine”, European Journal of Health Law, vol. 16, núm. 3, 2009, pp. 249-269; Dorbeck-Jung, B. y Chowdhury, N., “Is the Medical Products Authorization Regulation Equipped to Cope with the Challenges of Nanomedicines?”, Law & Policy, 33 (2), 2011; Duvall, M. N. et al., “Navigating FDA's Approach to Approval of Nanoparticle-Based Drugs and Devices”, Nanotechnology Law & Business, 8, 2011; Gispert, I., op. cit.; Ley Federal de Alimentos, Medicamentos y Cosméticos de Estados Unidos (FFDCA); Regulation 1394/2007/EC.
| Unión Europea | Estados Unidos de América |
|---|---|
| Normatividad | |
| Legislación farmacéutica - vols. 1 y 5, Normas sobre medicamentos de la Unión Europea. | |
| Órgano a cargo | |
| Agencia Europea de Medicinas (European Agency Medicine, EMA). | Agencia de Medicamentos y Alimentos (Food and Drugs Administration, FDA). |
| Órgano descentralizado de la UE. A cargo del proceso de autorización centralizado, realizan un análisis de riesgos/beneficios. | |
| Comités pertenecientes a EMA | Comités pertenecientes a FDA |
| Committee For Medicinal Products for Human Use (CHMP). Establecido por la Regulation No. 726/2004. Reconoce que los nanoproductos podrían abarcar los límites regulatorios entre productos sanitarios y dispositivos médicos. | Center of Drugs Evaluation an Research (CDER). Garantizar la seguridad y eficacia de los medicamentos, y disponibilidad de éstos. Regula medicamentos con y sin receta médica (“over-the-counter”), incluyendo terapias biológicas y medicamentos genéricos. Desarrolla la base de datos de productos con nanopartículas. |
| Scientific Committee on Emergingand Newly Identified Health Risk (SCENIHR). | Center of Biologics Evaluation and Research (CBER). Regula los productos biológicos y relacionados, incluyendo sangre, vacunas, alergénicos, tejidos y terapias celulares y genéticas. Revisión de nuevos productos biológicos y nuevas indicaciones de productos ya aprobados, la cual requiere la evaluación de datos científicos y clínicos presentados por los fabricantes para determinar si el producto cumple con los estándares de la CBER para su aprobación. |
| Innovation Task Force (ITF) - grupo multidisciplinario con competencias científicas, regulatorias y legales. Organización en grupos especializados: productos de terapia celular, productos de terapia genética, nanomedicinas, genómica, productos de combinación de dispositivos médicos y productos sanitarios. | Center for Devices and Radiological Health (CDRH). Asegurar que pacientes y proveedores tengan acceso oportuno y permanente a los productos que emiten radiación y sanitarios que sean seguros, eficaces y de alta calidad. Algunos de sus objetivos son mejorar los ensayos clínicos y encontrar un equilibrio adecuado en la recolección de datos de pre y poscomercialización. |
| Órgano a cargo | |
| Comités pertenecientes a EMA | Comités pertenecientes a FDA |
| Committee for Advanced Therapies (CAT) - procedimiento pequeñas y medianas empresas. Analiza datos de calidad y no clínicos de ATMPs en fase de desarrollo. Funciona como herramienta para iniciar negociaciones con grandes farmacéuticas o atraer inversionistas. | Office of Combination Products (OCP) - Oficina creada en 2002 por la FDA. Tarea de asignar productos de combinación (medicamentos, dispositivos y productos biológicos) a un centro en particular. Para determinar la competencia se hace una evaluación caso por caso con base en el modo de acción primaria del producto (“primary mode of action”). |
| Medicamento | |
| Definición | |
| 2004/27 Art. 1), «2), a) toda sustancia o combinación de sustancias que se presente como poseedora de propiedades para el tratamiento o prevención de enfermedades en seres humanos, o b) toda sustancia o combinación de sustancias que pueda usarse en o administrarse a seres humanos, con el fin de restaurar, corregir o modificar las funciones fisiológicas ejerciendo una acción farmacológica, inmunológica o metabólica, o de establecer un diagnóstico médico. | FFDCA § 201(h), 21 U.S.C. § 321(g)(1). Artículos reconocidos como tal por la farmacopea estadounidense; artículos para el diagnóstico, cura, mitigación, tratamiento o prevención de enfermedades en el hombre o animal, o artículos destinados a afectar la estructura o función del hombre o animal. |
| Normatividad específica a medicamentos | |
| Directiva 2004/27 - Medicamentos para uso humano. | FFDCA Capítulo V - Medicamentos y Aparatos § 351-360n. |
| Regulation No. 726/2004 setting down the Community procedures for the authorization and supervision of medicinal products. Art. 3. Define el alcance y la elegilibilidad de las solicitudes de evaluación bajo un marco de procedimiento centralizado (alcance: obligatorio, opcional, genérico/híbrido). | |
| Producto sanitario/dispositivos médicos/aparatos médicos | |
| Definición | |
| 2007/47 Art. 1), a), i), “a) ‹‹producto sanitario››: cualquier instrumento, dispositivo, equipo, programa informático, material u otro artículo, utilizado solo o en combinación, junto con cualquier accesorio, incluidos los programas informáticos destinados por su fabricante a finalidades específicas de diagnóstico y/o terapia y que intervengan en su buen funcionamiento, destinado por el fabricante a ser utilizado en seres humanos...”. | FFDCA § 201(h), 21 U.S.C. § 321(h) - Instrumento, aparato, implemento, máquina, artefacto, implante, reactivo in vitro, u otro artículo similar o relacionado, incluyendo una parte componente o accesorio. |
| Normatividad específica a productos sanitarios/dispositivos médicos/aparatos médicos | |
| Directive 2007/47/EC. Enmienda: Directive 90/385/ EEC - The Active Implantable Medical Device, Directive 93/42/EEC - The Medical Device (MDD) and Directive 98/8/EC - The Biocidal Products. | |
| Directive 98/79/EC - The In Vitro Diagnostic Medical Device Directive. | |
| Clasificación de productos sanitarios/aparatos médicos/ dispositivos médicos | |
| Clasificación de los productos sanitarios (Art. 9 MDD y Anexo IX): I. Productos no invasivos, o productos invasivos uso pasajero; IIa. Productos no invasivos destinados a la conducción o almacenamiento de sangre, fluidos o tejidos, o productos invasivos uso a corto plazo; IIb. Productos no invasivos destinados a modificar la composición biológica o química de la sangre u tejidos, o productos invasivos o implantables de uso prolongado; III. Productos invasivos tipo quirúrgico en sistema circulatorio central, sistema nervioso central, o con efecto biológico, o con modificaciones químicas en el organismo. | Clasificación de aparatos médicos (FFDCA § 513[a][1], 21 U.S.C. § 360c[a][1][A]): clase I - aquellos aparatos donde los controles generales, especificados en la FFDCA, son suficientes para ofrecer una garantía razonable sobre su seguridad y eficacia; clase II - aquellos aparatos que requieren de controles especiales para ofrecer una garantía razonable sobre su seguridad y eficacia; clase III - aquellos aparatos para los que no hay información suficiente para establecer que tanto controles generales como especiales ofrecen garantías razonables sobre su seguridad y eficacia. |
| Clases I y II presentan una ventaja comercial. En la mayoría de las circunstancias, los dispositivos en estas clases pueden solicitar una “notificación previa a la co-mercialización”, lo cual puede acelerar la entrada del dispositivo al mercado. | |
| Directive 2002/98/EC - On blood and blood products. | |
| Directive 2004/23/EC - On donation, procurement, testing, processing, preservation, as well as to manufactured products derived from human tissues and cells. | |
| Regulation 1394/2007/EC on Advanced Therapy Medicinal Products (ATMPs) - Nuevos productos médicos para uso humano basado en terapia genética, terapia celular somática o ingeniería tisular. | |
Existen dos conflictos principales con los regímenes normativos actuales. Por un lado, la diferente regulación de sustancias químicas y productos o dispositivos médicos no garantiza la seguridad ni la eficacia para el caso de nanomateriales, nanopartículas, nanofármacos y otros materiales a nanoescala. En muchos casos, las investigaciones y los resultados de éstas incluyen aspectos que podrían ser cubiertos por ambas regulaciones, además de que existen productos químicos de exportación e importación a nanoescala que no son supervisados o no cumplen con ninguna evaluación de seguridad o riesgos.44
Por otro lado, la categorización de los nanoproductos en medicamentos y productos sanitarios impacta sobre la naturaleza del proceso de aprobación tanto de la EMA como de la FDA.45 Así, los productos combinados, es decir, aquellos que como productos sanitarios administran o contienen fármacos, retan los límites entre las categorías regulatorias; por ejemplo, un dispositivo médico que funciona como transporte y, a su vez, se encarga de liberar y administrar el fármaco. Por lo tanto, esta categorización puede ocasionar confusiones, complicaciones o hasta vacíos legales graves.
De acuerdo con Bowman y Hogde, los marcos normativos vigentes en materia de NT contienen importantes vacíos o fisuras visibles, las cuales se magnifican en el marco internacional;46 por tal motivo, corresponde al gobierno asegurar una completa y eficaz regulación. Sin embargo, Bowman y Hodge concluyen que la evidencia científica no es suficiente para establecer un marco normativo en un corto plazo.47 De igual manera, Gispert afirma que para enfrentar los riesgos en salud y en medio ambiente es necesario un enfoque normativo incremental y no un régimen específico,48 aunque esto no implica que no sea necesario un marco normativo completo a largo plazo. De esta forma, es indispensable que la tecnología se desarrolle más y que tanto la industria como el gobierno revalúen los marcos a partir de la nueva evidencia científica y las preocupaciones sociales.49
El gobierno estadounidense se inclina por un marco normativo voluntario o coregulatorio; sin embargo, es poco probable que en el corto plazo se implemente un régimen normativo para nanomateriales.50 Por otro lado, la Unión Europea busca diseñar un marco normativo que (1) facilite el comercio, armonizando la regulación vigente aplicable, y (2) evite, prevenga o revele los posibles riesgos a la salud y al medio ambiente.
VConclusionesEs necesario considerar que a causa de las características físicas y químicas de las NTs es fundamental diseñar un marco regulatorio propio y específico. En un primer apartado se discutió la importancia de definir nanotecnología, nanomaterial, nanopartícula, etcétera, específicamente en el contexto jurídico. Posteriormente, se abordaron distintas propuestas teóricas que señalan la forma de un marco normativo. La constante entre las distintas posturas teóricas es el reto que enfrenta una regulación para mantener un equilibrio adecuado entre el fomento a la innovación y la creatividad contra el mantenimiento de la salud y la seguridad pública.
Las posturas teóricas pueden mostrar una visión de una regulación adecuada. Éstas van desde una regulación progresiva (enfoque dinámico), una integración de la colaboración pública (enfoque programático), una incorporación del aspecto ético (enfoque socioético), hasta una regulación escalonada (enfoque gradual). Ahora bien, si se toman cada uno de los aspectos que hacen únicos a estos cuatro enfoques teóricos y se aplican al diseño de un mismo marco normativo; es decir, una regulación gradual y dinámica que considere la opinión pública y que ésta participe en el proceso del diseño, y tomando en cuenta los aspectos y conflictos sociales, éticos y ambientales, en esencia los cuatro enfoques pueden ser incluyentes entre sí.
Después se analizó el marco normativo de la Unión Europea, los Estados Unidos y la ISO. Se comparó la normatividad respectiva de cada región o país para el caso de sustancias químicas, y en cuya reglamentación se norman sustancias en escala nanométrica. Asimismo, se consideraron el órgano regulatorio y las características intrínsecas de éste, los antecedentes o historia de la norma, y las diferencias que existen entre las tres normas. Por último, se mencionan los órganos encargados de regular los aspectos médicos, tanto en la Unión Europea como en los Estados Unidos, en relación con las NTs.
Se concluye que la regulación actual en materia de sustancias químicas es limitada en materia de NTs, debido a que hay procesos que no se abordan o supuestos que no aplican para las NTs, como es el caso del tonelaje en sustancias químicas menores a cien toneladas. Además, la separación en la regulación de sustancias químicas y medicamentos y productos sanitarios puede magnificar los vacíos legales existentes.
Igualmente, para el caso de la nanomedicina y las NTs, la categorización en medicamentos y aparatos médicos resulta inapropiada, ya que existen productos de combinación que pueden estar en ambas clasificaciones. Así pues, es posible hacer una separación o tomar una decisión basada en estudios caso por caso; sin embargo, según el avance que tengan la nanomedicina y sus aplicaciones, se darán más casos de productos de combinación y será necesaria una normatividad propia.
Candidato a doctor, Unidad en Estudios del Desarrollo de la Universidad Autónoma de Zacatecas.
Collingridge, D., The Social Control of Technology, Frances Pinter, 1980.
NNI, “National Nanotechnology Initiative”, 2013, disponible en: http://www.nano.gov.
OECD, “Regulatory Frameworks for Nanotechnology in Foods and Medical Products: Summary Results of a Survey Activity”, OECD. Science, Technology and Industry Policy Papers, núm. 4, 2013.
European Commission, Towards a European Strategy for Nanotechnology, Luxemburgo, European Commission, 2004.
Recomendación de la Comisión Europea de 18 de octubre de 2011 relativa a la definición de nanomaterial (2011/696/UE).
Rauscher, H. et al., Towards a Review of the EC Recommendation for a Definition of the Term “Nanomaterial”, Part 3, Italia, Publications Office of the European Union, 2015, disponible en: http://bookshop.europa.eu/uri?target=EUB:NOTICE:LBNA27240:EN:HTML (fecha de consulta: 29 de mayo de 2016).
National Institutes of Health, “National Cancer Institute”, 2013; NNI, op. cit.
Ramachandra M., Rayasa S., “Challenges and Emerging Issues in Patenting Nano-medicines”, en Souto, Eliana B. (ed.), Patenting Nanomedicines, Berlín-Heidelberg, Springer Berlin Heidelberg, 2012.
Puthli, S. et al., “Intellectual Property and Nanopharmaceuticals”, en Souto, Eliana B. (ed.), Patenting Nanomedicines, Berlín-Heidelberg, Springer Berlin Heidelberg, 2012.
Davies, J. C., Managing the Effects of Nanotechnology, Woodrow Wilson International Center for Scholars, Project on Emerging Nanotechnologies, 2006.
Khushf, G. y Siegel, R. A., “What Is Unique about Nanomedicine? The Significance of Mesoscale”, Journal of Law, Medicine & Ethics, 2012.
Davies, J. C., Oversight of Next-Generation Nanotechnology, Woodrow Wilson International Center for Scholars, Project on Emerging Nanotechnologies, 2009, p. 31.
Moor, J. H., “Why We Need Better Ethics for Emerging Technologies”, Ethics and Information Technology, 7 (3), 2005, pp. 111-119.
Idem.
Rollins, K., “Nanobiotechnology Regulation: a Proposal for Self-Regulation with Limited Oversight”, Nanotechnology, 6, verano de 2009.
Munir, Abu Bakar y Yasin, Siti Hajar Mohd, “Nanotechnology in Healthcare: Are Existing Laws Adequate?”, European Journal of Health Law, vol. 14, 2007, p. 261.
Ramachandran, Gurumurthy et al., “Recommendations for Oversight of Nanobiotechnology: Dynamic Oversight for Complex and Convergent Technology”, Journal of Nanoparticle Research, 13 (4), 2011, pp. 1345-1371.
Guerra, G., “European Regulatory Issues in Nanomedicine”, NanoEthics, 2 (1), 2008, pp. 87-97.
Marchant, G. E. y Sylvester, D. J., “Transnational Models for Regulation of Nano-technology”, Journal of Law, Medicine & Ethics, 2006.
Bowman, D. M. y Hodge, G. A., “Nanotechnology «Down Under»: Getting on Top of Regulatory Matters”, Nanotechnology Law & Business, junio de 2007.
Roco, M. C. y Bainbridge, W. S., “Societal Implications of Nanoscience and Nanotechnology: Maximizing Human Benefit”, Journal of Nanoparticle Research, vol. 7, núm. 1, 2005, pp. 1-13.
Foladori, G. et al., “Two Dimensions of the Ethical Problems Related to Nanotech-nology”, NanoEthics, vol. 3, núm. 2, agosto de 2009, pp. 121-127.
Viseu, A. y Maguire, H., “Integrating and Enacting «Social and Ethical Issues» in Nanotechnology Practices”, NanoEthics, vol. 6, núm. 3, 2012, p. 207.
Bruce, Donald, “Ethical and Social Issues in Nanobiotechnologies: Nano2Life Provides a European Ethical «Think Tank» for Research in Biology at the Nanoscale”, EMBO Reports, 2006, pp. 754-758.
Bennett, I. y Sarewitz, D., “Too Little, Too Late? Research Policies on the Societal Implications of Nanotechnology in the United States”, Science as Culture, 15 (4), 2006, pp. 309-325.
Wolf, Susan M. y Jones, Cortney M., “Designing Oversight for Nanomedicine Research in Human Subjects: Systematic Analysis of Exceptional Oversight for Emerging Technologies”, Journal of Nanoparticle Research, 13 (4), 2011, pp. 1449-1465.
Chau, Chi-Fai et al., “The Development of Regulations for Food Nanotechnology”, Trends in Food Science & Technology, vol. 18, núm. 5, mayo de 2007, p. 269; Ramachandran, Gurumurthy et al., op. cit.
Munir, Abu Bakar y Yasin, Siti Hajar Mohd, op. cit.
Roco, M. C. y Bainbridge, W. S., “Societal Implications...”, op. cit., p. 2.
Allhoff, F., “The Coming Era of Nanomedicine”, The American Journal of Bioethics, 9 (10), 2009, p. 8.
Roco, M. C. y Bainbridge, W. S., “Societal Implications...”, op. cit.
Idem.
Viseu, A. y Maguire, H., op. cit.
Sandler, R., “Nanomedicine and Nanomedical Ethics”, The American Journal of Bioethics, 9 (10), 2009, p. 17.
Allhoff, F., op. cit.
Idem.
Sandler, R., op. cit.
Brahic, Catherine y Dickson, David, “Helping the Poor: The Real Challenge of Nanotech”, SciDev.Net, febrero de 2005.
Justo-Hanani, R. y Tamar, D., “The Role of the State in Regulatory Policy for Nanomaterials Risk: Analyzing the Expansion of State-Centric Rulemaking in EU and US Chemicals Policies”, Research Policy, 43 (1), 2014, p. 176 (traducción propia).
Azoulay, D., “Just out of Reach”, The Center for International Environmental Law, 2012.
Guerra, G., op. cit.
Elaboración propia a partir de Justo-Hanani, R. y Tamar, D., op. cit.; Gispert, I., “Overview of Nanomedicines Regulation in the European Union”, Frontiers of Nanoscience, vol. 4 (1), 2012, pp. 487-507; ISO, “International Organization for Standardization”, 2014.
Elaboración propia a partir de Anatol, R. et al., “Continuing to Strengthen FDA's Science Approach to Emerging Technologies”, Nanomedicine: Nanotechnology, Biology and Medicine, 9 (5), 2013, pp. 594-599; Directive 90/385/EEC; Directive 93/42/EEC; Directive 98/8/EC; Directive 98/79/EC; Directive 2002/98/EC; Directive 2004/23/EC; Directive 2004/27/EC; Directive 2007/47/EC; D'Silva, J. y Van Calster, G., “Taking Temperature. A Review of European Union Regulation in Nanomedicine”, European Journal of Health Law, vol. 16, núm. 3, 2009, pp. 249-269; Dorbeck-Jung, B. y Chowdhury, N., “Is the Medical Products Authorization Regulation Equipped to Cope with the Challenges of Nanomedicines?”, Law & Policy, 33 (2), 2011; Duvall, M. N. et al., “Navigating FDA's Approach to Approval of Nanoparticle-Based Drugs and Devices”, Nanotechnology Law & Business, 8, 2011; Gispert, I., op. cit.; Ley Federal de Alimentos, Medicamentos y Cosméticos de Estados Unidos (FFDCA); Regulation 1394/2007/EC.
Bowman, D. M. y Hodge, G. A., “Nanotechnology «Down Under»...”, op. cit.
Duvall, M. N. et al., op. cit.
Bowman, D. M. y Hodge, G. A., “A Small Matter of Regulation: an International Review of Nanotechnology Regulation”, The Columbia Science and Technology Law Review, vol. VIII, 2007.
Bowman, D. M. y Hodge, G. A., “Nanotechnology «Down Under»...”, op. cit.