





In this work, we studied the aptitude to sintering green bodies using γ-Al2O3 transition alumina as raw powder. We focused on the influence of the heating rate on densification and microstructural evolution. Phase transformations from transition alumina γ→δ→θ→α-Al2O3 were studied by in situ X-rays diffraction from the ambient to 1200°C. XRD patterns revealed coexistence of various phase transformations during the heating cycle. DTA and dilatometry results showed that low heating rate leads to a significant reduction of the temperature of the α-Al2O3 alumina formation. Around 1190, 1217 and 1240°C were found when using 5, 10 and 20°C/min of heating rate, respectively. The activation energy for θ-Al2O3→α-Al2O3 transformation calculated by Kissinger and JMA equations using dilatometry method were 464.29 and 488.79kJ/mol, respectively and by DTA method were 450.72 and 475.49kJ/mol, respectively. In addition, the sintering of the green bodies with low heating rate promotes the rearrangement of the grains during θ-Al2O3→α-Al2O3 transformation, enhancing the relative density to 95% and preventing the development of a vermicular structure.
En este trabajo, se ha estudiado la capacidad de sinterización de muestras en verde a partir de γ-Al2O3 de transición en forma de polvo. El trabajo se ha focalizado en la influencia de la velocidad de calentamiento sobre la densificación y la evolución microestructural. Las transformaciones de fase de alúminas de transición γ→δ→θ→α-Al2O3 se han estudiado in situ mediante Difracción de Rayos X (DRX) desde temperatura ambiente hasta 1.200°C. Los diagramas de XRD han revelado la coexistencia de diversas transformaciones de fase durante el ciclo de calentamiento. Los Análisis Térmicos Diferenciales (ATD) realizados y los datos de dilatometría han mostrado que velocidades de calentamiento bajas conducen a una reducción significativa de la temperatura de formación de α-Al2O3. Detectándose alrededor de 1.190, 1.217 y 1.240°C cuando se utilizan 5, 10 y 20°C/min como velocidad de calentamiento, respectivamente. La Energía de Activación para la transformación θ-Al2O3→α-Al2O3 calculada mediante las ecuaciones de Kissinger y JMA usando métodos dilatometricos han sido 464,29 y 488,79kJ/mol, respectivamente, y mediante ATD 450,72 y 475,49kJ/mol, respectivamente. Además, la sinterización con velocidad de calentamiento baja promueve la reorganización de los granos durante la transformación θ-Al2O3 → α-Al2O3, el aumento de la densidad relativa al 95% y la prevención del desarrollo de una estructura vermicular.
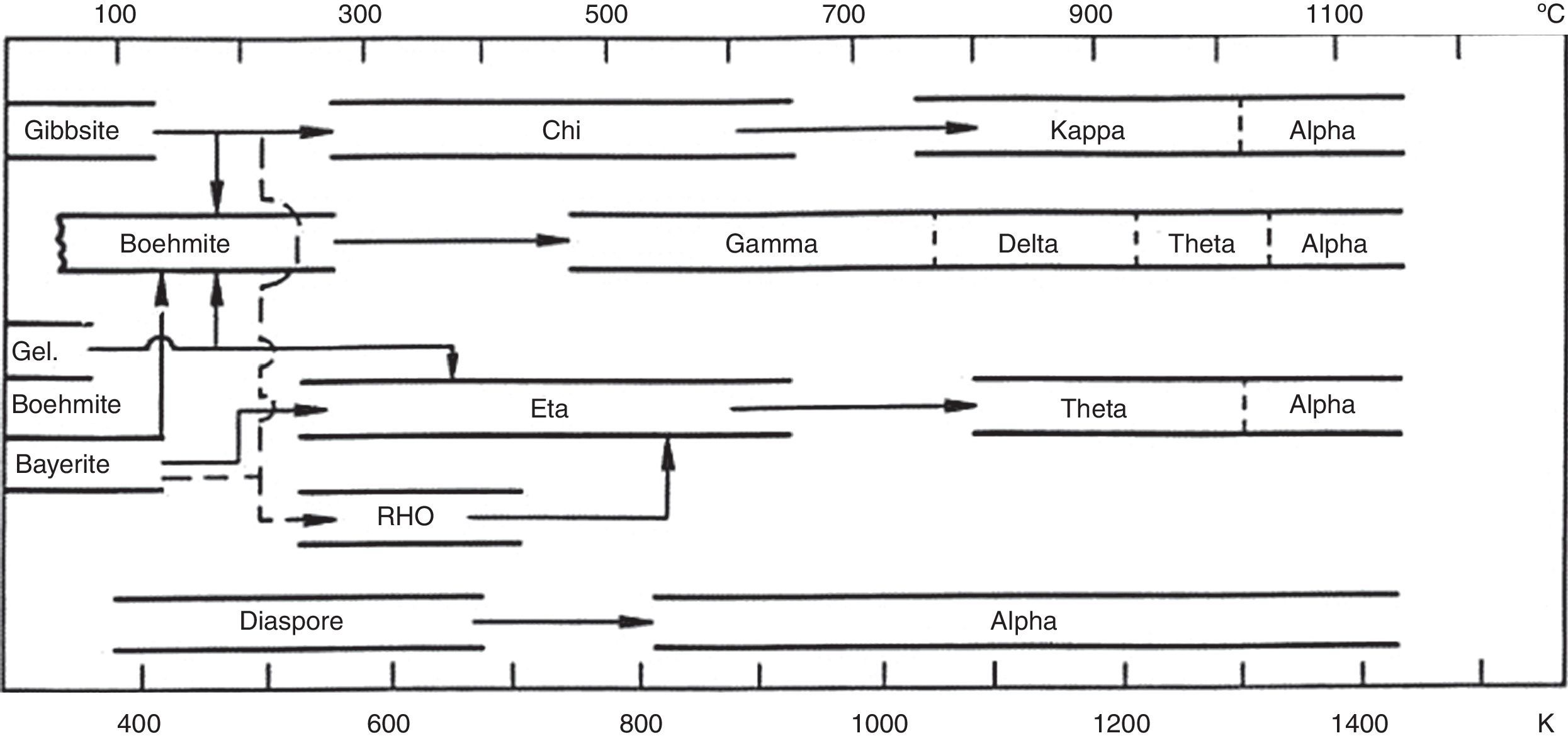
Due to its interesting physico-chemical and mechanical properties, alumina is one of the most used technical ceramics as structure parts, applied in medical use (orthopaedic implants) and electronics [1–5]. To obtain dense alumina products, a perfect processing control from powder synthesis to sintering [6,7] is required. Gibbsite (Al(OH)3) and boehmite (AlOOH) are the most used precursors for the preparation of α-Al2O3. During the heat treatment, aluminium hydroxides transform into transition alumina in the form of metastable structure before ending as α-Al2O3 thermodynamic stable phase (Fig. 1) [8,9]. Boehmite transforms into γ-Al2O3 transition alumina under a temperature range of 500–550°C with a departure of structural water [10]. The transformation of γ-Al2O3 monoclinic phase (d=3.56gcm−3) to α-Al2O3 hexagonal phase (d=3.98gcm−3) is accompanied by a volume reduction of about 10% causing a large density increase [11–14]. The transformation of θ-Al2O3 transition alumina to α-Al2O3 takes place in the temperature range 1050–1200°C. This transformation occurs through a nucleation and grains growth mechanism [15–18] and is influenced by several parameters such as grain size [19], chemical composition [20] and heating rate [21]. The θ-Al2O3→α-Al2O3 transformation is very significant for the sintering process and consequently for the control of microstructure [22]. According to the sintering conditions, the formation of crystalline α-Al2O3 can produce a vermicular microstructure form consisting of a wide pores network, which develops during the α-Al2O3 transformation [23,24]. In addition, it is difficult to study with precision the transformation of transition alumina phases due to their instability and high reactivity [21]. Different studies showed that a high energy milling influences the phase transformation from transition alumina to α-Al2O3 stable phase [25–29]. Specific diagrams defining the pathway of phase transformations were established according to the heating temperature and particle grain size [10,30]. Some studies were conducted using coarse and fine grained gibbsite in order to examine the transformation paths using in situ X-rays diffraction [19]. Others concern the gibbsite and boehmite to α-Al2O3 phase transformation cases. They noticed that small α-Al2O3 crystallites of spherical form are obtained by boehmite transformation and larger ones in plate form from gibbsite [31]. The raw powder and the milling mode are therefore fundamentally important for the sintering process and the densification. Many techniques are used to improve the densification process like α-Al2O3 seeds [32,33] and the alumina doping agents [34].
![Transformation sequences of gibbsite (Al(OH)3) to α-A12O3[10].](https://static.elsevier.es/multimedia/03663175/0000005600000002/v1_201704290021/S0366317516300899/v1_201704290021/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNd3TCEZuNxhudow/+3Hrh9WJc+mtnYSSPL9K6mfMWVj1qgNs5/3ZsuL6PqEb/cSmTokCoGyLIUoo8OmfI6fr7UqJ6egmqO6l7DVV1MtoPC4PYAcKkTk6/WrOzsUhfx2yDpOleDgZFmjrjxeXdbxsQw3NtAfSB84uhJ9OseJuMB3TZao/f0cMPzzuEZ+j59LjFRGu1+zDpSQ3qx2fwafuOWLvI+qxJOfGX2dAdUmTucyg+uOzsFX6miIss/F1XWUmAI= "Transformation sequences of gibbsite (Al(OH)3) to α-A12O3[10].")
Transformation sequences of gibbsite (Al(OH)3) to α-A12O3[10].
In this work we used γ-Al2O3 transition alumina as raw powder to study the densification process and the microstructure of sintered samples using various heating rates. The γ-Al2O3 to α-Al2O3 transformation was characterized in situ by XRD, and the activation energy accompanying the θ-Al2O3 to α-Al2O3 transformation was evaluated.
Experimental proceduresThe used raw powder is γ-Al2O3 transition alumina obtained by boehmite calcinations at 600°C (boehmite being obtained by partial dehydration of gibbsite at 450°C). The chemical composition expressed in weight % is given as follows: Al2O3(88.34%), SiO2(3.86%), F(0.89%), Na2O(0.34%), CaO2(0.18%), Fe2O3(0.096%) and structural water(6%). The powder was attrition milled during 3h using alumina balls of 2mm diameter. A 0.25% mass ratio of DARVAN C as a dispersant and 1% of duramax as a bonder were added. During the milling process, the pH was adjusted at 10.4 [35]. After milling, the suspensions were completely dried at 110°C. The powder was sieved to obtain aggregates smaller than 45μm. In order to highlight the phase transformations, the thermal behaviour of the powder was studied using an analyser SETERAM TGA 92 allowing a differential thermal analysis (DTA). Thermal cycles were recorded from ambient up to 1600°C with three heating rates (5, 10 and 20°C/min). A powder mass of nearly 40mg was used in all cases. The analysis by X-rays diffraction was carried out using Bruker D8 Advance diffractometer working at high temperatures. Green samples thermal shrinking was also studied between room temperature and 1300°C. Dilatometry tests were carried out using a NETZSCHDIL402C dilatometer. The green compacts were initially shaped by uniaxial pressing then by cold isostatic pressing at 300MPa. The samples were sintered at 1700°C for 2h. The final densities were determined by Archimedes's method using distilled water. The SEM micrographs were obtained using a JOEL 840A scanning electronic microscope. A 20kV voltage and a working distance between 10 and 15mm were used. The gold metalized polished surfaces were observed after a thermal treatment at 1600°C during 1h.
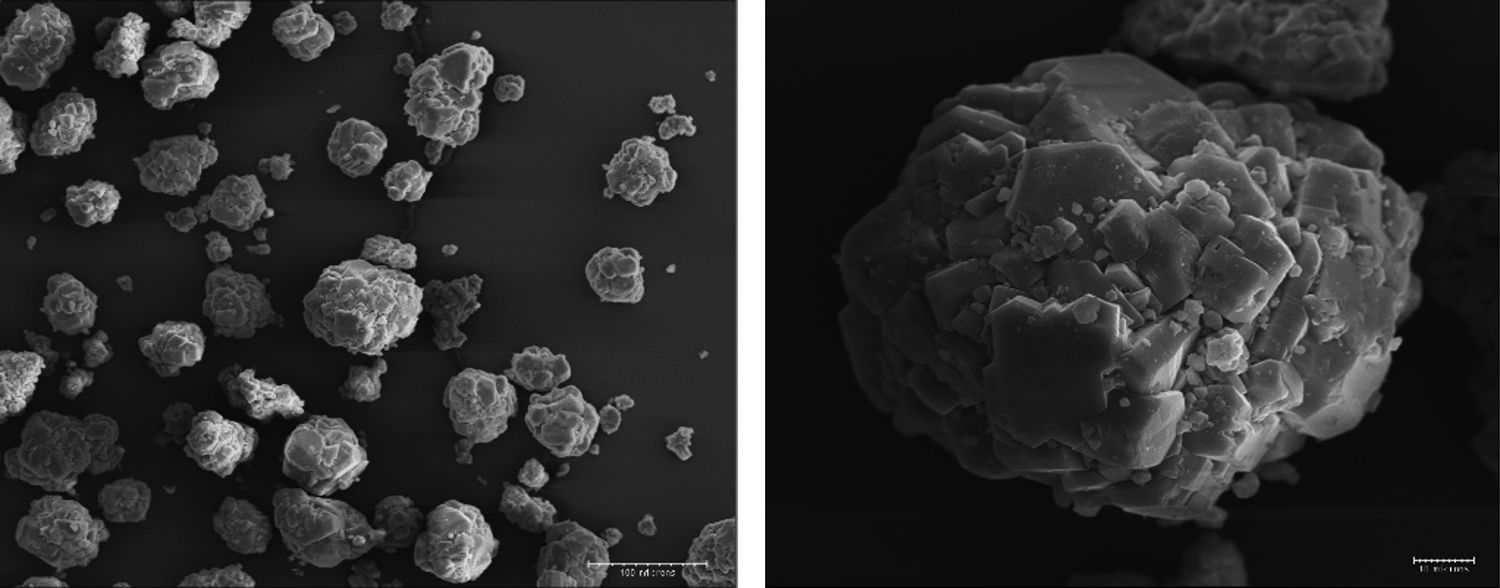
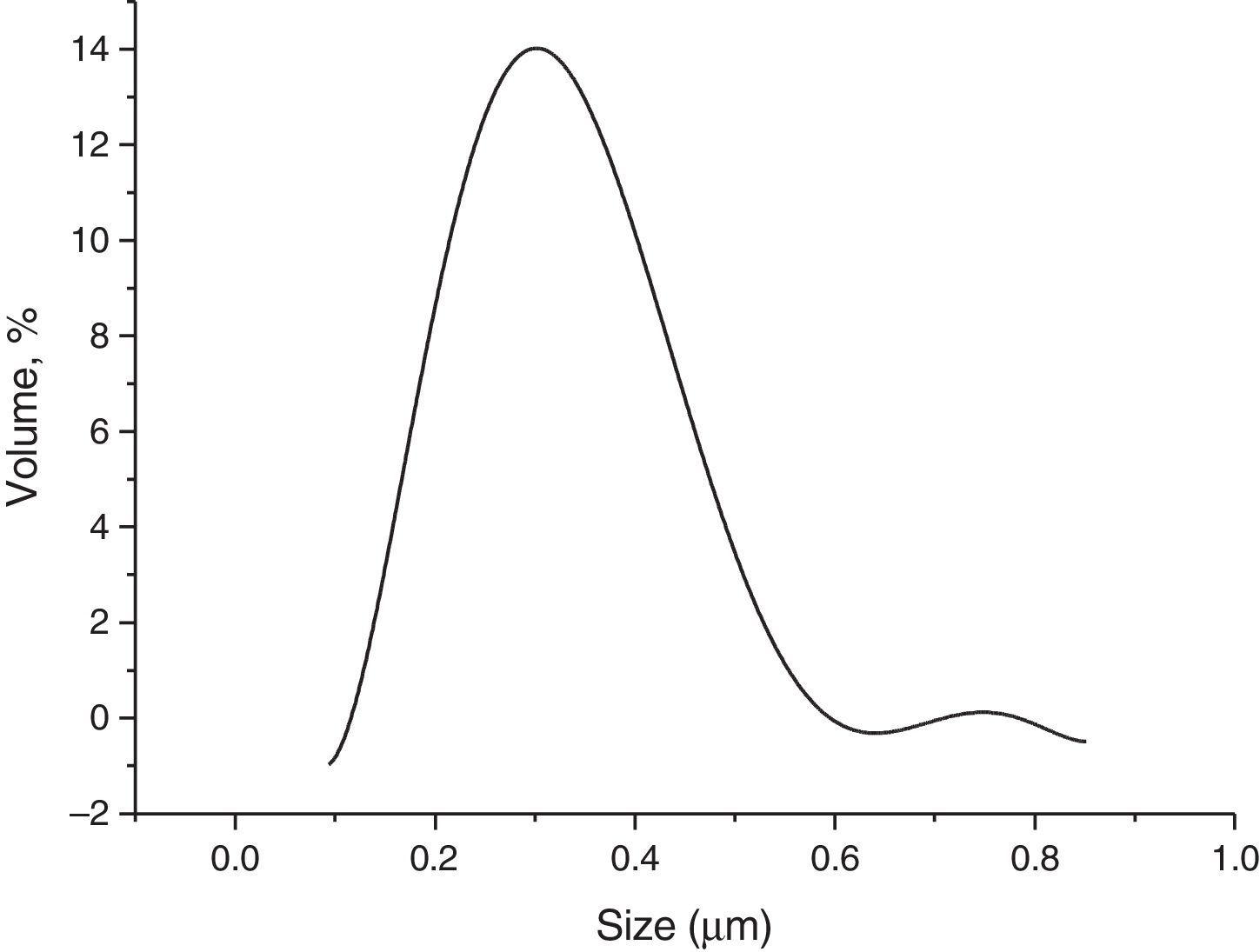
Results and discussionsRaw powderThe morphology of the used raw boehmite is illustrated in Fig. 2. It consists of agglomerates of around 75μm. The raw powder was attrition milled reducing its mean diameter (D50) down to 0.29μm (Fig. 3). The absolute density of the milled powder measured with helium pycnometer was 3.08g/cm3. To obtain the γ-Al2O3 transition alumina phase, the milled boehmite was heated at 600°C in order to completely eliminate the structural water according to established methods [30].
Differential thermal analysis

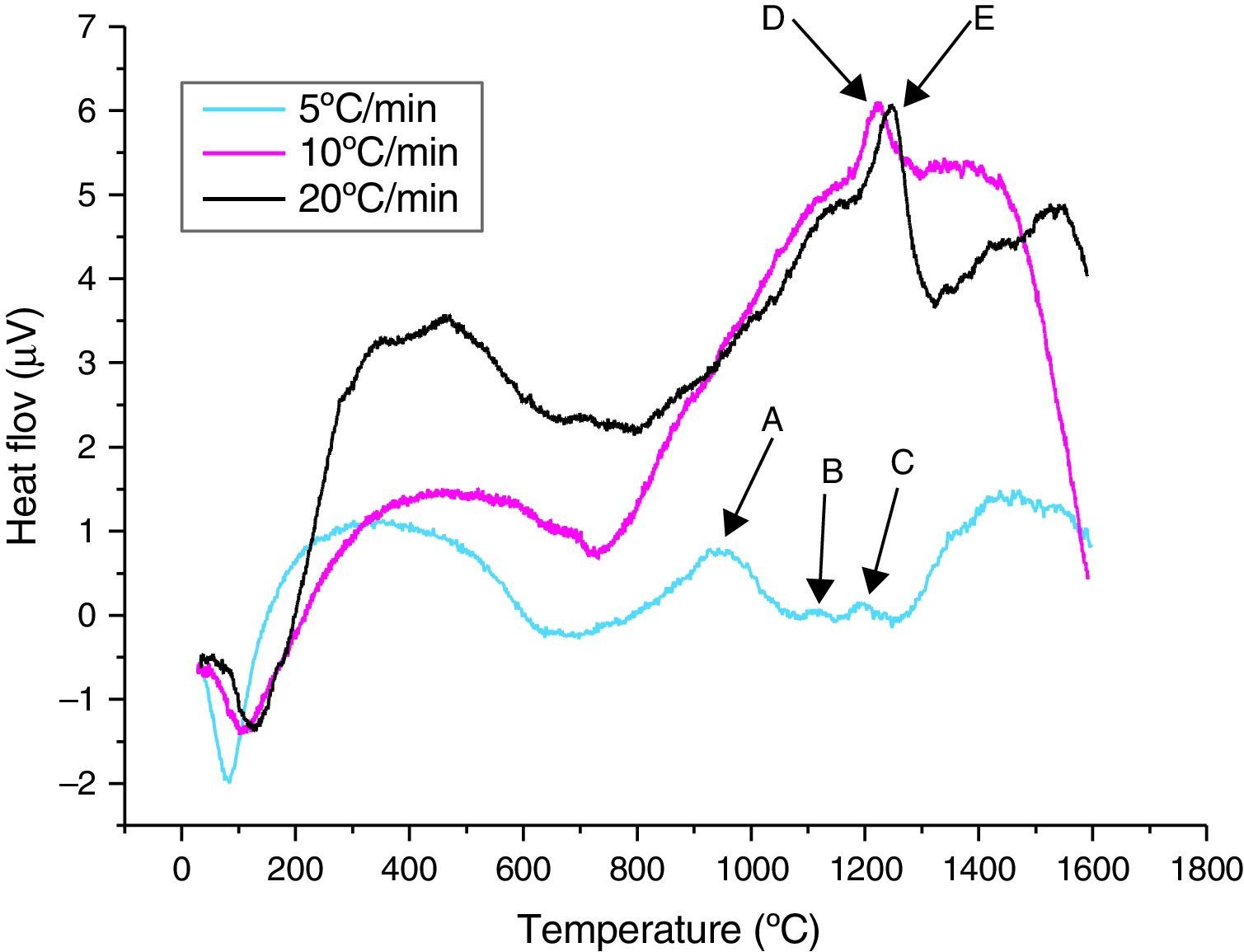
The thermal behaviour (DTA) of the γ-Al2O3 transition alumina powder, for different heating rates, is illustrated in Fig. 4. During the heating cycle between ambient and 1600°C, the curves revealed that:
- -
For a 5°C/min heating rate, the points A, B and C, three exothermic peaks of transition alumina transformation phases are observed. The first peak at around 800°C is related to the transformation of γ-Al2O3 to the δ-Al2O3, the second at around 1080°C is due to the δ-Al2O3→θ-Al2O3 transformation and finally the third peak at 1150°C to the θ-Al2O3→α-Al2O3 phase transformation.
- -
For 10 and 20°C/min heating rates, only the θ-Al2O3→α-Al2O3 phase transformation appeared. They respectively start at 1217°C (Point E) and 1240°C (point F). Our results correspond to Weffer and Misra diagram [10]. A shift in the θ-Al2O3→α-Al2O3 transformation peak for the three heating rates is also observed. The lowest heating rate reduces the temperature of α-Al2O3 formation. The transformation seems more progressive, with a larger and less intense peak than for higher heating rates.

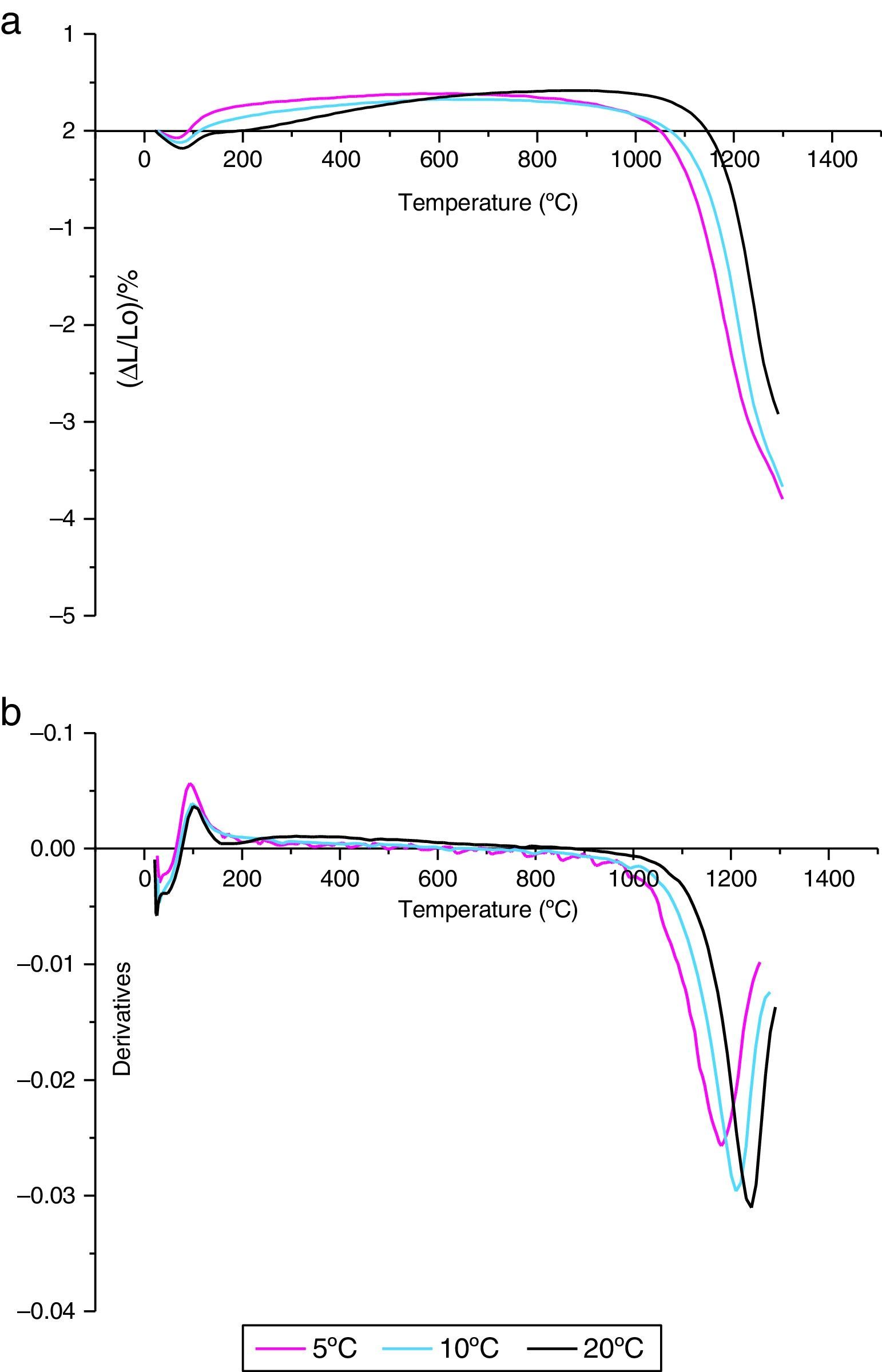
The shrinkage curves and their derivatives of the pressed samples are shown on (Fig. 5). They indicate two notable variations along the heating cycle. The samples shrinkage was disrupted by a small expansion indicating the θ-Al2O3→α-Al2O3 phase transformation at 1056, 1082 and 1140°C for the heating rate of 5, 10 and 20°C/min respectively. The lowest heating rate promotes a maximum transformation rate at 1150°C. This type of expansion was observed by different authors under the influence of alpha alumina doping seeds, sample compaction pressure rates and doping agents [8,34]. From the crystallographic point of view, the θ-Al2O3→α-Al2O3 phase transformation occurs by crystallographic structure change (oxygen anion moving positions) from cubic face centred towards a compact hexagonal structure.
 and their derivatives (b) of the γ-Al2O3 transition alumina samples heated up to 1300°C for different heating rates.")
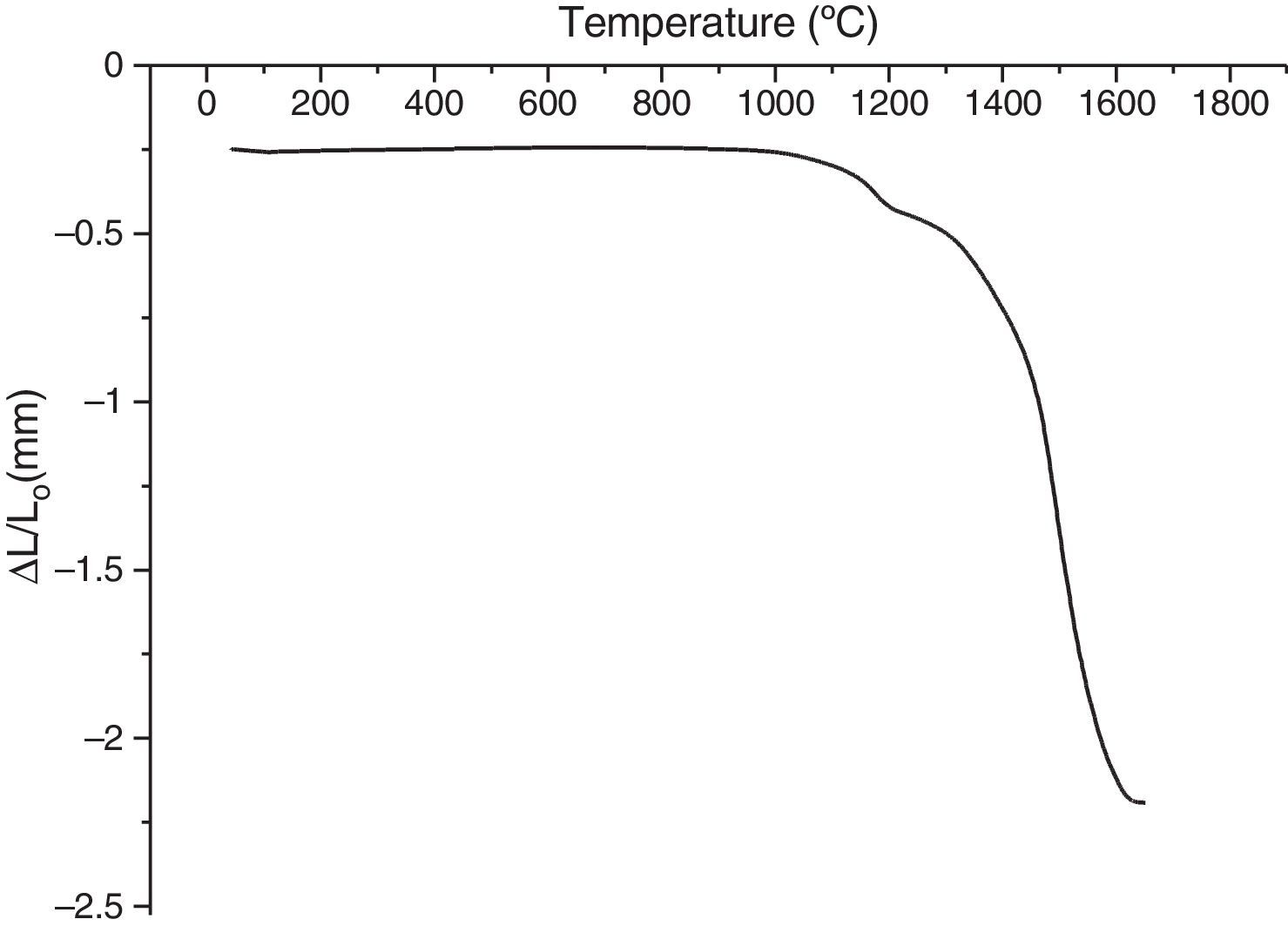
This transformation takes place by nucleation and growth mechanisms. It occurs with high activation energy depending on the heating rate [16–36]. In such transformation, the activation energy is mostly used in the nucleation process. This induces the temperatures shifts at the beginning of the θ-Al2O3→α-Al2O3 phase transformation caused by the crystallographic reorganizations mechanisms [37]. In order to complete and evaluate the sintering process, a dilatometric cycle of γ-Al2O3 transition alumina of pressed green sample from ambient to 1700°C with a 5°C/min heating rate was made (Fig. 6). This test enables us to make a shrinkage process study. No volume variation was observed at the beginning of sintering process. This can be explained by the fact that the transition alumina phase transformations are characterized by structural changes without shrinkage. The θ-Al2O3→α-Al2O3 phase transformation initiates at 1050°C and reaches its maximum at 1192°C.
DRX in situ
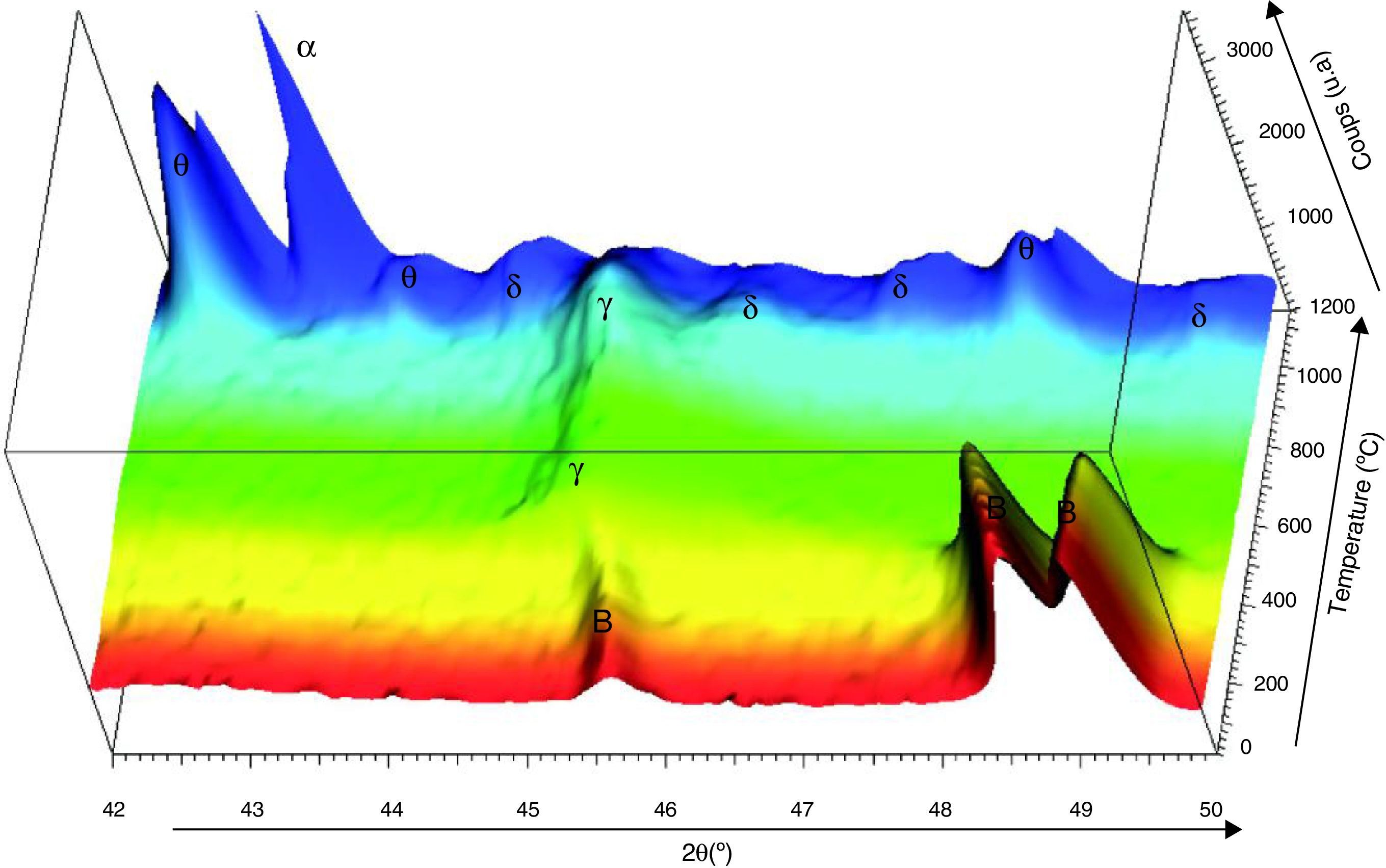
Fig. 7 shows the XRD patterns carried in situ from ambient to 1200°C on boehmite attrition milled raw powder. The choice of this monohydrate comes from the fact that γ-Al2O3 transition alumina is produced from the dehydration of fine boehmite [10]. The XRD data were collected on a Bruker D8 Advance diffractometer with angular sweeping from 2θ=42° until 2θ=50° at 0.1°/min. The phases were identified by comparison with the JCPDS tables (γ-Al2O3: ICDD 50-741, δ-Al2O3: ICDD 16-394, θ-Al2O3: ICDD 23-1009, α-Al2O3: ICDD 46-1212). From the ambient to 450°C, the diffractograms shows that the observed peaks correspond to crystalline boehmite. Boehmite is completely transformed to γ-Al2O3 in the temperature range 550 to 850°C. The γ-Al2O3 and δ-Al2O3 phases coexist up to 950°C. Beyond this temperature, the θ-Al2O3 starts to form progressively. The spectra of those “pseudo-amorphous” δ-Al2O3 and θ-Al2O3 phases decrease gradually with formation of the α-Al2O3 stable phase. Beyond 1200°C, only the α-Al2O3 is observed.
Activation energy
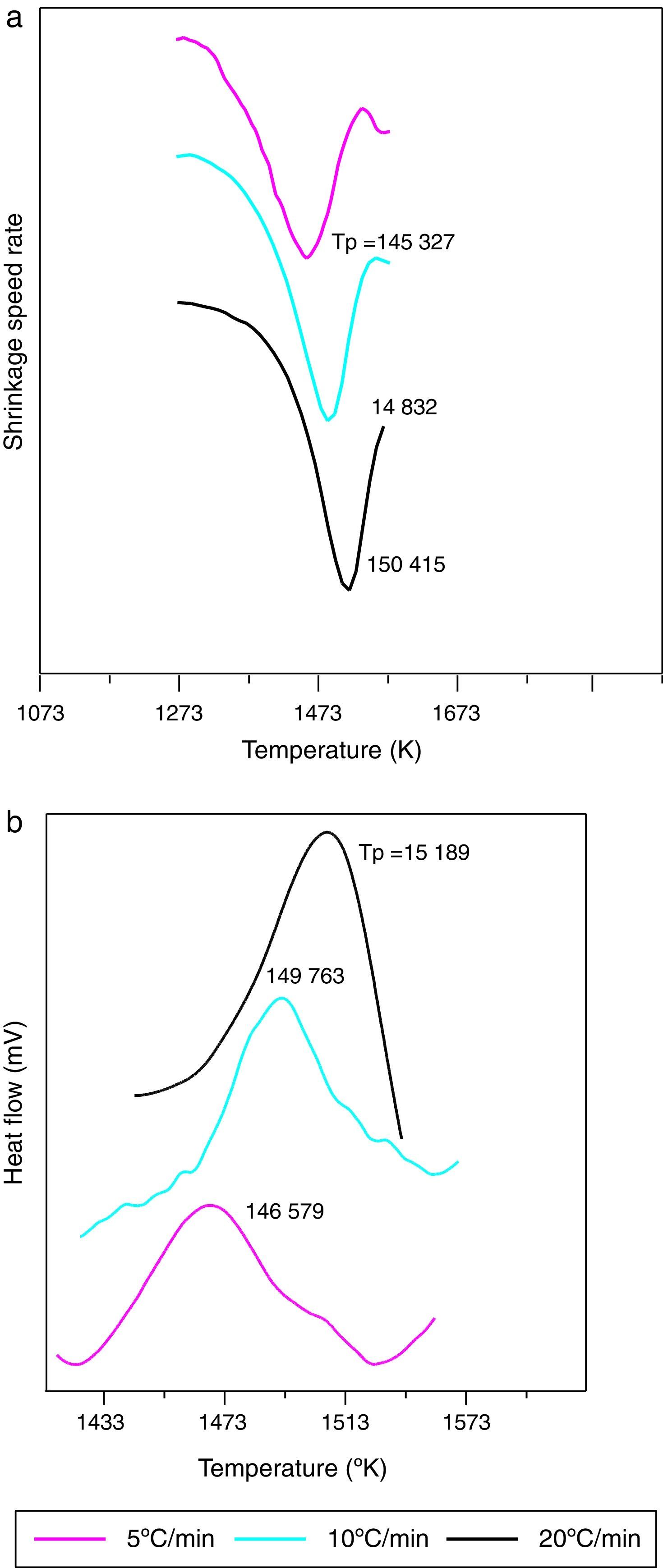
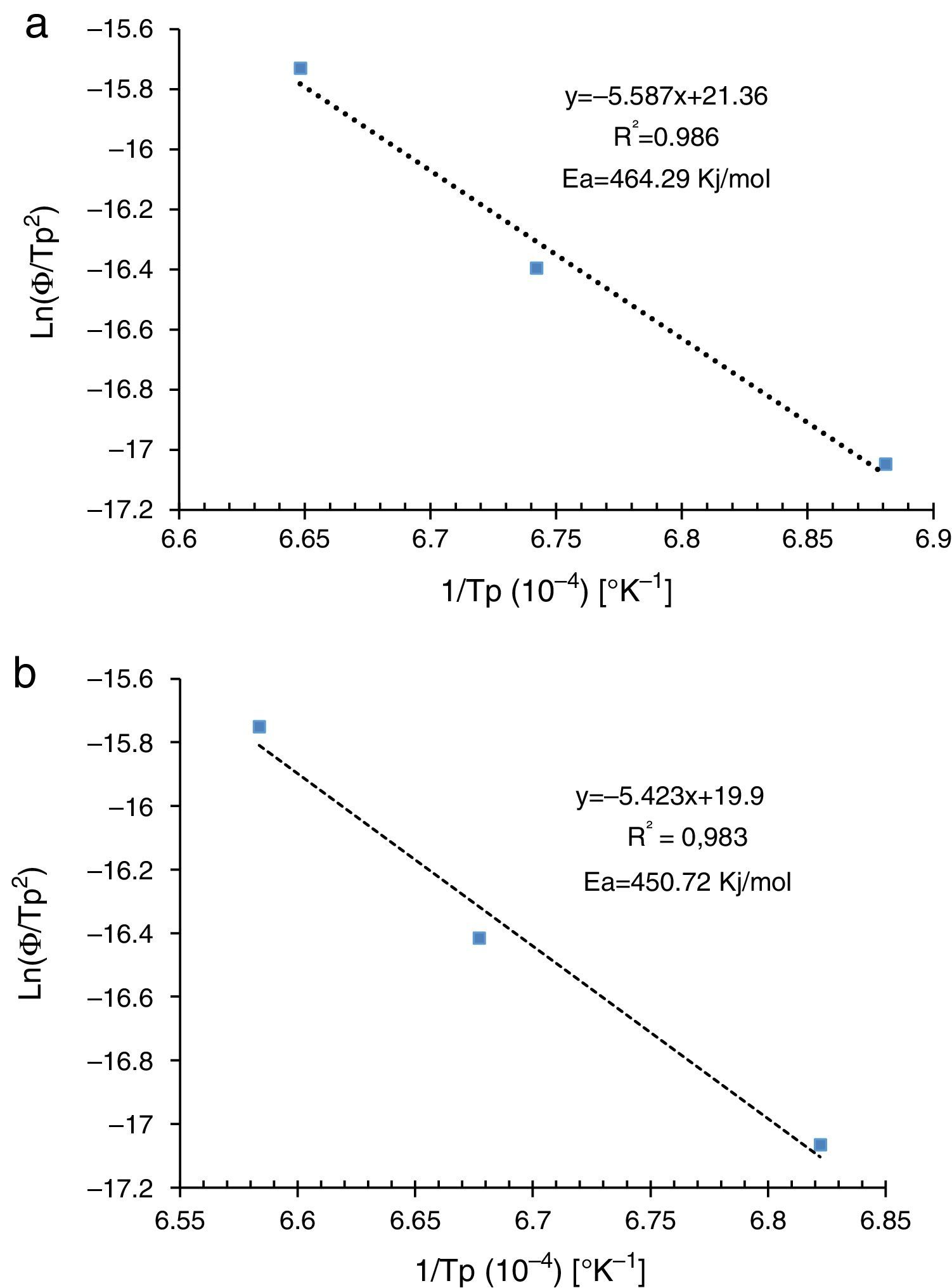
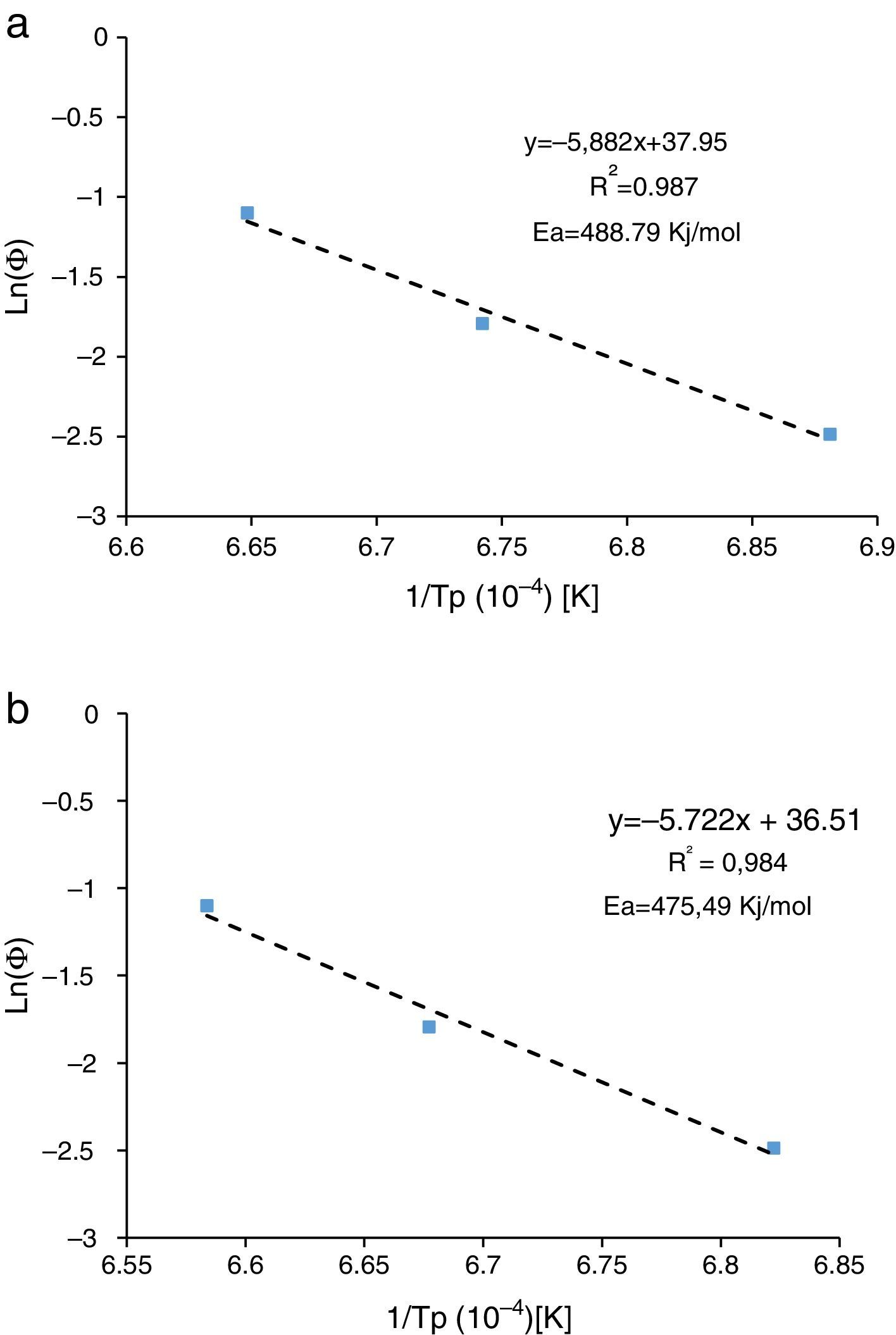
Dilatometry and DTA tests were undertaken to evaluate the activation energy of θ-Al2O3→α-Al2O3 transformation. The activation energy (Ea) values were obtained from transformation temperature peak measurements [20,38] using the three heating rates (Fig. 8). The activation energy values for θ-Al2O3→α-Al2O3 transformation were estimated by Kissinger and (JMA) methods [39] using the following Eqs. (1) and (2), according to the peak temperature Tp at the different heating rates:
where ‘ϕ’ is the heating rate (K/s), Tp is the temperature corresponding to the maximum of θ→α-Al2O3 transformation peak (K), Ea is the activation energy of the θ→α-Al2O3 transformation (kJ/mol) and R is the ideal gas constant (8.314J/molK).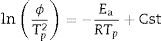
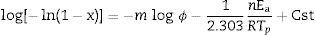
 and DTA curves (b) at various heating rates.")
The relation characterizing the variable x:
where AT is the area of the exothermic peak in the DTA curve at temperature T and A0 is the area under the peak.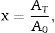
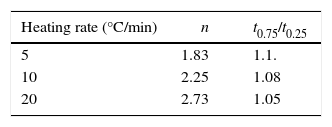
The plots obtained in Figs. 9 and 10, (ln(ϕ/Tp2) vs 1/Tp) and (ln (ϕ) vs 1/Tp) respectively, are characterized by straight lines with different slopes Ea at different heating rates. The obtained activation energy value by dilatometry was about (464kJ/mol). The one obtained by DTA is about 450kJ/mol. These close values remain lower than those reported by Ma. Krell and Wang (517kJ/mol) [38,39]. According to these results, the γ-Al2O3 transition alumina raw powder attrition milled contributes to lowering activation energy for the formation of the stable α alumina phase. On the other hand, the value of the exhibitor Avrami parameter [40,41], which reflects the transformation rate by germination and growth, is defined by (Eq. (3)) from the endothermic shape of crystallization obtained by DTA. The morphology of the crystal growth can be obtained at the transformation ratios (75% and 25%) [42].
where ΔT is the width of the crystallization peak at half maximum value. The values of the Avrami constants are listed in Table 1. They increase from 1.83 to 2.73 with increasing heating rate. The average values of t0.75/t0.25 for each heating rate are also listed in Table 1. These values are very close suggesting that the growth morphology is the same at the different heating rates.Microstructure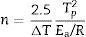
 versus 1/Tp (a) by dilatometry curves, (b) by DTA curves.")
 by dilatometry curves, (b) by DTA curves.")
The SEM micrographs of green bodies’ sintered samples at 1700°C are shown in Fig. 10. The γ-Al2O3 samples heated with a heating rate 20°C/min had a porous structure with 85% of relative density and 1.6μm grain mean size (Fig. 10a). For the sample fired with a 10°C/min heating rate, a microstructure improvement was noticed. The grain boundaries are quite apparent with important intergranular porosity. The relative density is 90% and the mean grains size is 1.54μm (Fig. 10b). On the other hand, the samples heated at 5°C/min had a dense and homogeneous microstructure with low residual porosity and 95% relative density and 3.2μm mean grain size (Fig. 10c). The observed high density and homogeneous crystal structure with clearly apparent grain boundaries are a caused by a well established particles rearrangement during the θ-Al2O3→α-Al2O3 transformation process (Fig. 11). The residual porosity in intra-granular parts was difficult to eliminate [19]. Otherwise, the presence of impurities in the starting alumina powder could be at the origin of liquid silicates formation. Generally, this glassy phase is located in grain boundaries and triple points. The formation of the amorphous phase facilitates alumina densification during sintering (Fig. 12).
, 10°C/min (b), and 5°C/min (c).")

The influence of the heating rate on the densification and microstructures of green bodies using γ-Al2O3 raw powder was studied during sintering. With a low heating rate (5°C/min), the DTA curves showed a γ-Al2O3→α-Al2O3 phase transformations. For higher heating rates (10 and 20°C/min), only the θ-Al2O3→α-Al2O3 phase transformation was detected. The dilatometry curves showed that the temperature at the beginning of the θ to α alumina transformation decreases with decreasing heating rates. The in situ XRD results for the boehmite raw powder used showed coexistence of different transition alumina during the phase transformations. The diffraction patterns in the temperature range (550–850°C) correspond to the γ-Al2O3 phase. At 950°C, this phase is still present and coexists with the δ-Al2O3 phase. Beyond this temperature, we observed the θ-Al2O3→α-Al2O3 phase transformation. The activation energy for this transformation was evaluated using Kissinger equation from the results obtained by both DTA and dilatometry techniques. The SEM micrographs showed that samples heated at a 5°C/min rate had a high relative density (95%) and homogeneous microstructure with low residual porosity.

