





En este estudio se han preparado un material cerámico denso, con una composición nominal (% en peso) de 40 Ca3(PO4) – 60 (SiO3)2, mediante sinterización en estado sólido de polvos finos de Ca3(PO4)2 y CaMg(SiO3)2 sintéticos. Los resultados obtenidos por DRX y microscopia electrónica de barrido de emisión de campo con microanálisis indican que los materiales obtenidos presentan una microestructura homogénea, con un tamaño de grano fino, compuesta por granos de diópsido (CaMg(SiO3)2) y whitlockita (β-Ca3(PO4)2ss) junto con muy pequeñas cantidades de apatita.
Los valores de tensión de fractura y el módulo de elasticidad del material optimizado son similares a los del hueso humano.
La bioactividad del material se ha evaluado experimentalmente estudiando la formación in vitro de apatita en suero fisiológico simulado. Con el objetivo de comprender los resultados obtenidos en los estudios in vitro se ha simulado la disolución de las diferentes fases presentes en el material en SFA mediante cálculos termodinámicos.
Durante el experimento in vitro en SFA los granos de whitlockita se disuelven más rápidamente que los de diópsido lo que origina una superficie porosa rica en diópsido. Posteriormente, tiene lugar la disolución del diópsido remanente en la superficie del material de β-Ca3(PO4)2-CaMg(SiO3)2 que, después de siete días en SFA, queda recubierta por una capa de apatita.
Se espera que este material biocerámico de β-Ca3(PO4) y CaMg(SiO3)2 sea útil para la fabricación de andamiajes para reparación ósea.
In this study, a dense bioactive ceramic, with nominal composition (wt.%) 40 Ca3(PO4)2–60 CaMg(SiO3)2, was prepared by solid state sintering of homogeneous compacted mixtures of fine synthetic Ca3(PO4)2 and CaMg(SiO3)2 powders. The results obtained by X-ray diffraction and field emission scanning electron microscopy with microanalysis indicate that the ceramic composite showed a fine grained and homogeneous microstructure consisting of diopside (CaMg(SiO3)2) and whitlockite (β-Ca3(PO4)2ss) grains with very small amounts of apatite.
The flexural strength and elastic modulus values of the composite are similar to those of cortical human bone.
Bioactivity was experimentally evaluated by examining in vitro apatite formation in simulated body fluid (SBF). In addition, a simulation of the dissolution properties of the different phases present in the material in SBF was carried out by thermodynamic calculations, with the purpose of understanding the in vitro results obtained.
The experimental results demonstrated that, during soaking in SBF, the grains of whitlockite dissolved preferentially than those of diopside, leaving a porous surface layer rich in diopside. Subsequently, partial dissolution of the remaining diopside occurred and the porous surface of the ceramic became coated by a bone-like apatite layer after 7 days in SBF.
This bioceramic containing β-Ca3(PO4)2 and CaMg(SiO3)2 is expected to be useful to fabricate scaffolds for bone repair.
Calcium phosphate based bioceramics have been in use in medicine and dentistry for 30 years. They are used in a wide range of applications, including coatings of implants, alveolar ridge augmentation, maxillofacial surgery and scaffolds for bone growth and as powders in total hip and knee surgery ([1]). In the last years many efforts in artificial bone engineering have involved the use of synthetically obtained tricalcium phosphate ([2]) (Ca3(PO4)2, TCP), as a bioresorbable compound in clinical use for bone-repair ([3], [4]); however the control of its bone-bonding ability, i.e. bioactivity, and bioresorbability is not easy ([5], [6], [7], [8]). On the other hand, Si and Mg have received great attention as substitutes in the Ca3(PO4)2 network, to improve osteogenesis, bioreabsorption rate, strength, and phase composition of the resulting bioceramics ([9]). Diopside (CaMg(SiO3)2, D) is a potential candidate as Mg- and Si-containing source for bioceramics because it has been found to be non-cytotoxic, biocompatible and bioactive in vitro (slightly in SBF, and fully in human parotid saliva) and in vivo ([10]). On the other hand, dense diopside ceramics show high mechanical strength and toughness, as compared to TCP (KIC ≈ 3.5 and 1.1 MPa m1/2 and σf ≈ 300 and 180 MPa for D and TCP ([11]), respectively; and low biodegradability ([12])). Diopside has also been reported to be able to closely bond to bone tissue when implanted in rabbits.
Control of the surface reactivity of bioactive ceramics to surrounding body fluids is essential in the design of novel bone-repairing materials, because apatite formation on bioactive materials is governed by the dissolution of constituents from the materials and the nucleation of hydroxyapatite on the resultant surface.
The combination of a resorbable ceramic compound as Ca3(PO4)2, with low strength, with a high strength bioactive one, such as CaMg(SiO3)2 would led to novel designs of bioactive and bioresorbable materials for use as bone regeneration materials. Moreover, bone formation will be stimulated by the presence of silicon and magnesium ([13], [14]).
Ashizuka et al. ([15]) reported that the glass-ceramics prepared from glasses with the eutectic composition (wt.%) of 38 Ca3(PO4)2-62 CaMg(SiO3)2 could reach flexural strength values of about 200 MPa. However the bioactivity of this material was not analysed by the authors. Kamitakara et al. ([16]) investigated the fundamental parameters of preparation of dense and porous glass ceramics by devitrification of the parent glass with eutectic composition of the system Ca3(PO4)2-CaMg(SiO3)2. These authors observed that both porous and dense diopside-β-tricalcium phosphate glass-ceramics show bioactivity and bioresorbability in a simulated body fluid.
In a previous work, Carrodeguas et al. ([17]) reported a preliminary study of the in vitro behaviour of dense ceramics of Ca3(PO4)2-CaMg(SiO3)2 (60:40 wt.%). These authors observed the formation of a bone-like apatite layer on the interface between the simulated body fluid and the material.
Recently Guerrero-Lecuona et al. ([18]) in an in vitro study about the influence of the structure and microstructure on the bioactive behaviour of a ceramic and a glass-ceramic with composition (wt.%) 45 CaO, 22 SiO2, 28 P2O5 and 5 MgO observed that the glass-ceramic dissolved faster, releasing large proportion of Ca and P ions to the liquid media.
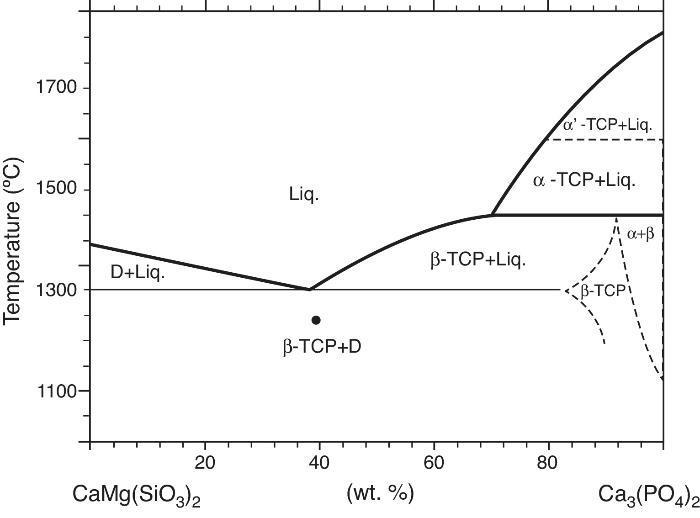
Phase relations in the Ca3(PO4)2-CaMg(SiO3)2 system were established by Sata ([19]) and revised by Carrodeguas et al. ([20]) (Fig. 1). Sata ([15]) reported a eutectic point located at 63 wt.% CaMg(SiO3)2 and 1300 ± 5 °C. In addition, the presence of important solid solutions of Ca and Si in the low temperature polymorph of Ca3(PO4)2 was reported by Carrodeguas et al. ([20]), while presence of solid solutions in CaMg(SiO3)2 has not been detected.
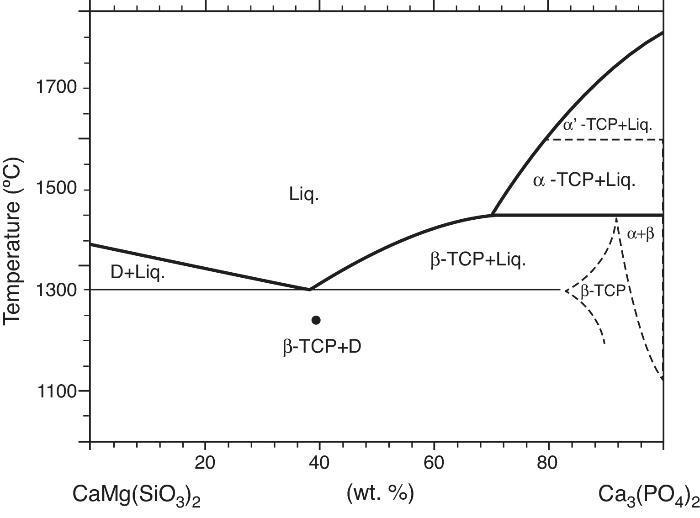
Fig. 1. Redrawn phase diagram of the Ca3(PO4)2-CaMg(SiO3)2 system according the present authors [19] .
This study aimed to investigate the preparation, by solid state sintering, of a biphasic composite in the system Ca3(PO4)2-CaMg(SiO3)2 (40:60 wt.%), and the evaluation of its microstructure, properties and potential as bioceramic for bone repair.
The studied composition, close to the eutectic of the pseudo-binary system Ca3(PO4)2-CaMg(SiO3)2, was formulated on the basis of the information supplied by the equilibrium relationships ([18], [21]). It is hypothesized that the bioceramic, being constituted by one bioactive and one resorbable phase will develop an in situ porous structure when submerged in SBF ([22]).
ExperimentalMaterial processingThe starting materials were synthetic Ca3(PO4)2 (TCP) and CaMg(SiO3)2 (D) prepared by solid state reaction.
Ca3(PO4)2 was prepared by solid state reaction between CaCO3 (with purity >99.5%, Merck, Darmstadt, Germany) and CaHPO4 (>99.0 wt.%, Panreac, Barcelona, Spain). Reactants were intimately mixed in an attrition mill during 45 min using isopropanol and 3 mm ∅ with yttrium partially stabilized zirconia (Y-PZS) balls. The resulting slurry was dried at 60 °C overnight. The dried cake was crushed and sieved through 100 μm mesh and the powder was heated at 1100 °C for 4 h. Finally the product was grinded in a tungsten carbide mill to a mean particle size of 1.9 μm, as measured by laser scattering (Master Sizer S; Malvern Instruments Ltd., Worcestershire, UK). The chemical analysis accomplished by X-ray fluorescence spectroscopy (XRF, Magi X; Phillips, the Netherlands) indicated that the Ca/P molar ratio is 1.485 implying that the powder is calcium-deficient (Ca/P <1.5) being the major impurity MgO (0.38 ± 0.02 wt.%; see Table 1).
Table 1. Chemical analysis and physical characteristics of the starting materials Ca3(PO4)2, and CaMg(SiO3)2.
| CaMg(SiO3)2 | Ca3(PO4)2 | |
| Chemical analysis (wt.%) | ||
| CaO | 26.8 ± 0.3 | 53.59 ± 0.03 |
| SiO2 | 54.8 ± 0.3 | 0.086 ± 0.005 |
| Al2O3 | 0.019 ± 0.005 | 0.056 ± 0.005 |
| MgO | 18.0 ± 0.3 | 0.38 ± 0.03 |
| Na2O | 0.061 ± 0.002 | 0.089 ± 0.002 |
| P2O5 | n.d. | 45.66 ± 0.05 |
| K2O | 0.013 ± 0.001 | n.d. |
| MnO2 | n.d. | 0.025 ± 0.003 |
| Fe2O3 | 0.045 ± 0.004 | 0.032 ± 0.004 |
| SrO | n.d. | 0.020 ± 0.003 |
| SO3 | n.d. | 0.065 ± 0.005 |
| Physical characteristics | ||
| Specific surface area (m2/g) | 1.5 | 1.4 |
| Average grain size (μm) | 1.90 | 2.30 |
| Density (g cm−3) | 3.23 | 2.88 |
Chemicals used in the synthesis of diopside were high-purity amorphous silica (99.50 wt.% min; Cerac Inc., Milwaukee, WI); CaCO3 (99.50 wt.% min, Merck, Darmstadt, Germany) and MgO (99.99 wt.% min, Merck, Darmstadt, Germany). Stoichiometric quantities of the starting powders were attrition milled with Y-PZS balls in isopropyl alcohol media, dried, isostatically pressed and heated in platinum crucibles at a rate of 5 °C min−1 up to 950 °C for 2 h. To ensure the homogeneity the pre-reacted CaMg(SiO3)2 was ground, isostatically pressed and reheated at 1300 °C to ensure chemical homogeneity. This procedure was repeated until CaMg(SiO3)2 was the only crystalline phase detected by X-ray diffraction (XRD). The obtained CaMg(SiO3)2 was crushed and milled in attrition mill (Y-PSZ balls, in isopropyl alcohol). The results of chemical analysis by X-ray fluorescence (XRF), for both materials, are shown in Table 1.
Subsequently, a stable aqueous slurry of the mixture of powders (40 wt.% Ca3(PO4)2–60 wt.% CaMg(SiO3)2) with 0.8 wt.% Dolapix CE 64 (Zschimmer & Schwarz, Lanstein, Germany) was attrition milled with Y-PSZ balls to an average particle size of 5 μm. The slurry was then dried and sieved and the powder was isostatically pressed at 200 MPa into plates (≈100 mm × 100 mm × 15 mm).
In order to establish the optimum sintering schedule, constant heating rate (10 °C min−1) experiments were done in a horizontal dilatometer (DI-24, Adamel-Lhomargy, France). The green plates were sintered in air at 1225, 1250 and 1275 °C/4 h using heating and cooling rates of 5 °C/min.
Specimens for characterization were diamond machined from the sintered plates.
Material characterizationThe bulk density of the sintered bodies was measured by the Archimedes method employing water.
The crystalline phases present in the sintered material were identified by X-ray diffraction (XRD) on powder samples with particle size under 125 μm. XRD patterns were recorded with a powder diffractometer D8 Advance (Bruker AXS GmbH, Karlsruhe, Germany) with copper anode (CuKα1 λ = 0.15418 nm) working at 40 kV and 40 mA. Measurements were done on samples rotating at 15 r.p.m. in the interval 2–90° (2θ), X-ray patterns were acquired with a step/size of 0.03° and time/step of 20 s.
The experimental diffraction patterns were compared to the standards of the Joint Committee on Powder Diffraction Standards (JCPDS) database reported for α-Ca3(PO4)2 (JCPDF #9-348), β-Ca3(PO4)2 (PDF #9-169), Ca10(PO4)6(OH)2 (PDF # 9-432) and CaMg(SiO3)2 (PDF#11-654).
The microstructure of the ceramic composite was studied on both fracture and polished surfaces using a Field Emission Scanning Electron Microscope (FE-SEM, Hitachi S-4700, Japan) with X-ray energy-dispersive spectrometry (CFE-SEM-EDS). Polishing was done with diamond suspension spray of grain size of 6, 3 and 1 μm. Polished surfaces were chemically etched with diluted CH3COOH (5% vol/2 s).
Rectangular bars of 5 mm × 5 mm × 50 mm were used for measuring the flexural strength and the elastic modulus. The surfaces of the machined specimens were polished with 6 μm diamond suspension and the tensile side surface in specimens used to measure the strength was mirror-polished to 3 μm diamond suspension.
The Young's modulus was determined from the resonance frequency of the bars in bending (GrindoSonic MK5, J. W. Lemmens, Belgium). The strength was determined in 3-point bending (span: 20 mm) at a crosshead speed of 0.5 mm/min. Given results for Young's modulus and strength are the average of 10 determinations and errors are the standard deviation.
SBF in vitro testIn vitro studies were made using the S imulated B ody F luid (SBF) immersion test proposed by Kokubo and Takadama ([23]). The Tris-buffered SBF No. 9 (Na+ 142.0, K+ 5.0, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3− 5.0, HPO42− 1.0 and SO42− 0.5 mol/m3), with ion concentrations nearly equal to that of human blood plasma was used.
Plates of the ceramic material (≈10 mm × 10 mm × 5 mm) were hanged with a nylon thread in polyethylene test tubes containing 100 mL of SBF at 36.5 ± 0.5 °C and pH = 7.25. The ratio volume of SBF/area of ceramic was equal to 0.5 cm3/mm2. The SBF was prepared and was replaced every two days. Plates were removed at 1, 2, 3 days and 1, 2 and 3 weeks of soaking time, gently washed with deionized water and acetone, and dried in air at room temperature.
The concentration of Ca, Mg, P and Si ions in the removed SBF was determined by means of inductively coupled plasma atomic emission spectroscopy (ICP-AES, Jarrel Ash Iris Advantage, USA). The pH of the SBF was monitored by mean of a pH-meter with an Ag/AgCl reference electrode.
The sample surfaces and the cross sections, before and after the exposure to the SBF, were examined (FE-SEM) and EDS elemental maps (Ca, Mg, Si and P), of the cross sections were collected.
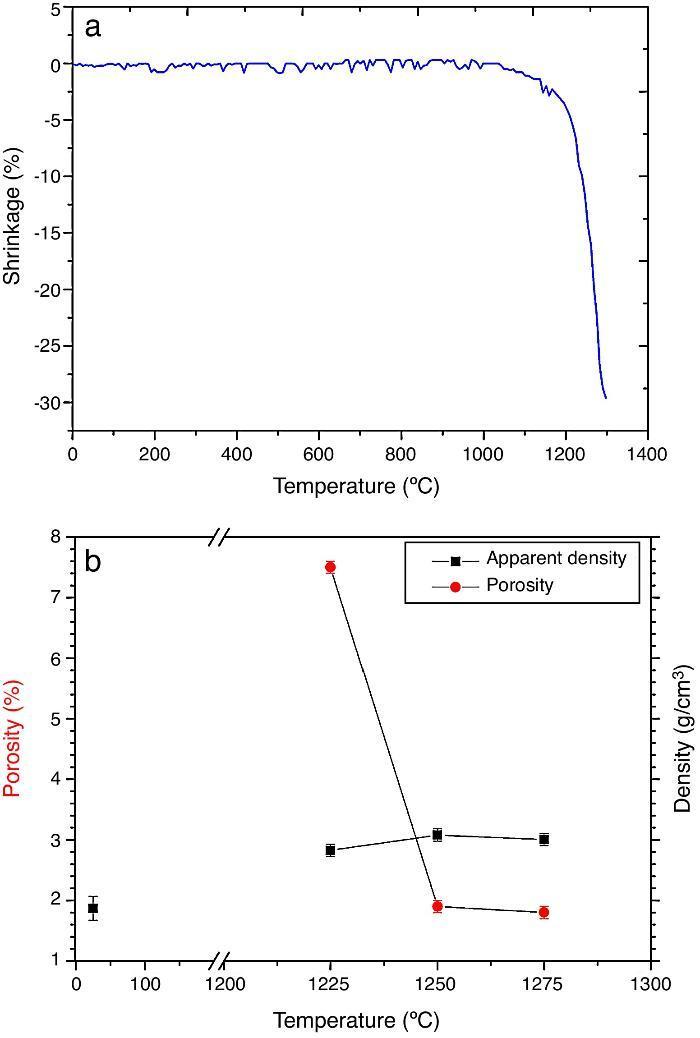
ResultsMaterial characterizationShrinkage recorded during the constant heating rate tests is shown in Fig. 2a. Maximum rate was reached at ≈1250 °C. In Fig. 2b the apparent density of sintered specimens is plotted versus the maximum sintering temperature. The maximum density was reached at 1250 °C.
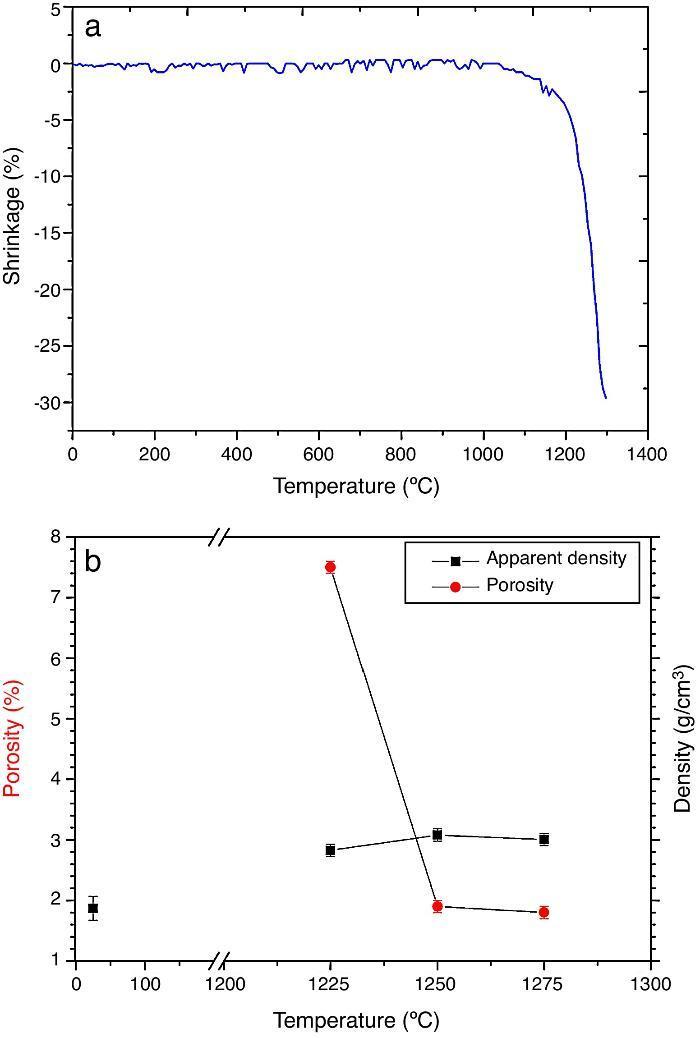
Fig. 2. Dilatometric analysis. (a) Constant heating rate sintering curve of the green compact specimen; (b) Apparent density and open porosity as a function of temperature for 4 h of heating. (Heating and cooling rate 5 °C × min−1).
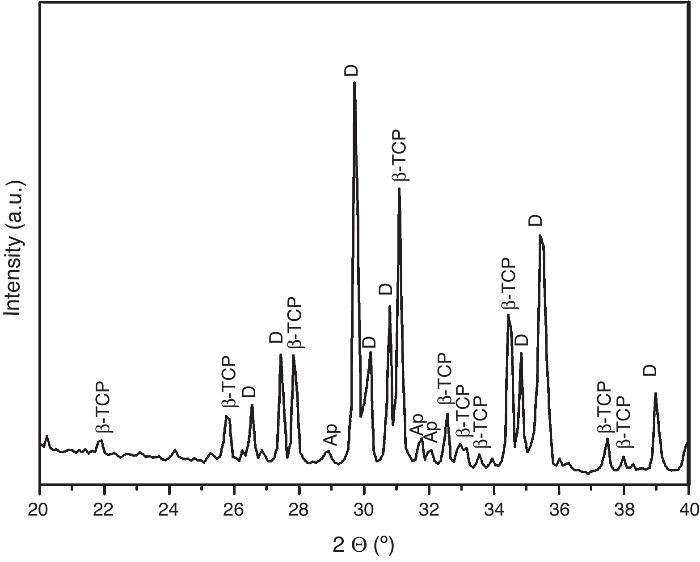
For the three sintering temperatures, the XRD patterns showed the presence of the same crystalline phases: D, β-TCP and traces of hydroxyapatite (Ap) (Fig. 3).
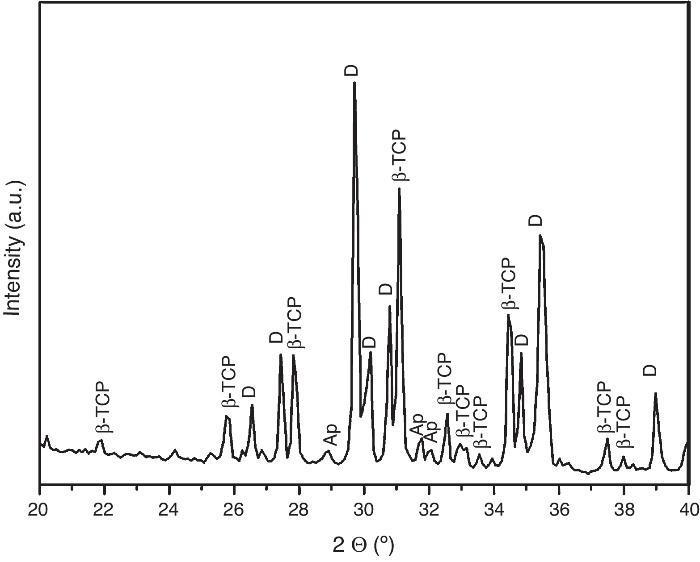
Fig. 3. Powder X-ray diffraction pattern of the specimen obtained by sintering at 1250 °C for 4 h. The different phases β-Ca3(PO4)2 (β-TCP), CaMg(SiO3)2 (D) and apatite (Ap) were identified.
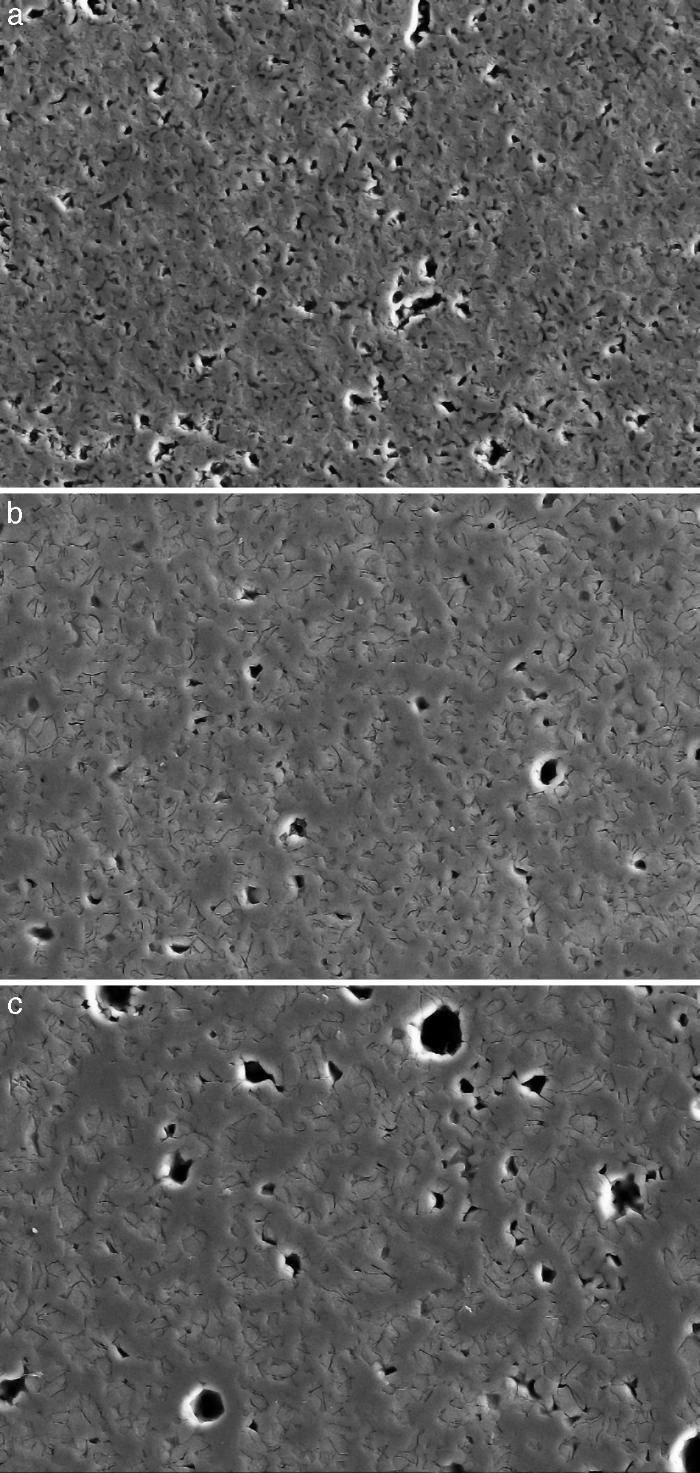
Fig. 4, Fig. 5 show the characteristic microstructural features of the sintered materials. The microstructure was constituted by the homogeneous distribution of two kinds of areas with extremely different responses to etching (Fig. 4). The strongly etched areas were highly microcracked and their composition can be associated to β-TCP. These areas were surrounded by an uncracked matrix of diopside, as was revealed by EDS microanalysis. The size of these areas increased with temperature (Fig. 4). Porosity decreased from 1200 to 1250 °C sintering temperature. The size of the pores increased sharply from ≈3 to 5 μm for the lowest sintering temperatures (1200 and 1250 °C) up to ≈10 μm for the material sintered at 1275 °C.
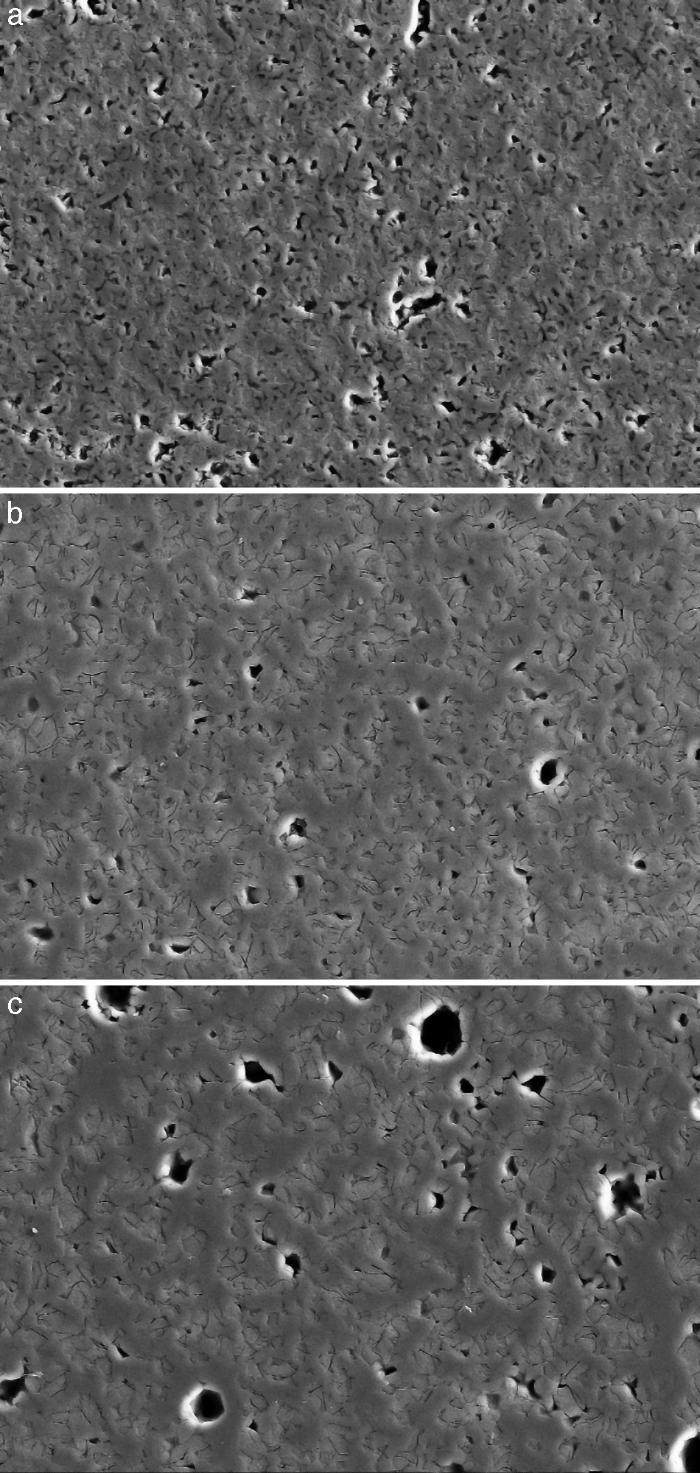
Fig. 4. Low magnification CFE-SEM microphotographs of the polished and chemically etched surfaces (5 vol.% acetic acid/2 s) of Ca3(PO4)2-CaMg(SiO3)2 samples sintered 4 h at: (a) 1225 °C; (b) 1250 °C and (c) 1275 °C.
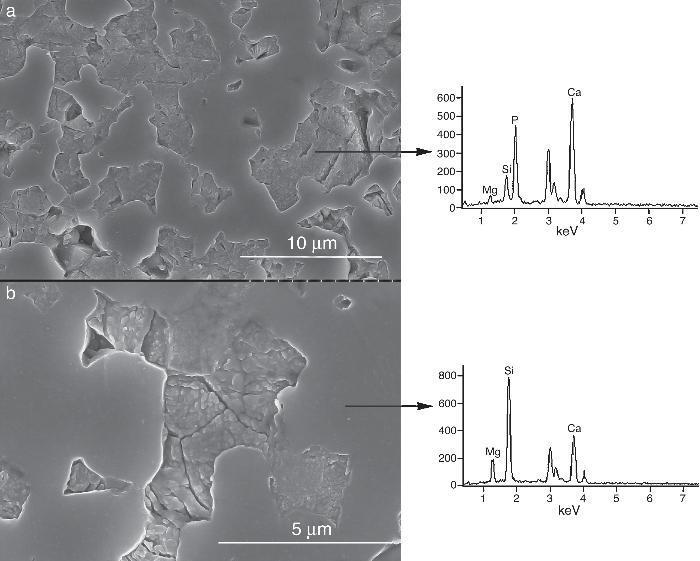
Fig. 5. CFE-SEM microphotographs of the sample sintered at 1250 °C/4 h and chemically etched with dilute acetic acid showing: (a) general view, and (b) detail. X-ray energy-dispersive spectrometry analysis of diopside, and tricalcium phosphate phases.
Fig. 5a shows the close up of the typical microstructure of the sample heated for 4 h at 1250 °C, in which it can be observed light grains of diopside, with sizes of ∼6 μm, and gray grains of tricalcium phosphate (3–4 microns) with several cracks, 3–4 μm long, in their grain boundaries The results of EDS semi-quantitative analysis of the tricalcium phosphate grains, confirms the presence of Mg and Si in solid solution in β-TCP. The small amounts of apatite identified by X-ray diffraction could not be identified by FE-SEM.
The volume fraction of each phase was determined by image analysis, obtaining 51 ± 4 vol.% for the ligth phase (D) and 49 ± 4 vol.% for the gray phase dissolved phase (tricalcium phosphate). These results are in agreement with the phase diagram data if taking into account the solid solution of Mg and Si in tricalcium phosphate at 1250 °C (48 vol.% TCP and 52 vol.% D).
As the material sintered at 1250 °C presented the lowest porosity and homogeneous and relatively fine microstructure, this thermal treatment was chosen as optimum and specimens sintered at 1250 °C were selected.
Mechanical propertiesTable 2 lists values for the flexural strength and elastic modulus obtained for the material sintered at 1250 °C/4 h, values for trabecular and cortical bone ([1]) and dense calcium phosphate ceramics are also included as reference ([13], [24]).
Table 2. Mechanical properties of various bioactive biomaterials.
| Property | Bone [1,4] | TCP [21] | D [11] | 40TCP-60D |
| Mineralogical composition (vol.%) | ||||
| Diopside | – | – | 100 | 51 ± 4 |
| Tricalcium phosphate | – | 100 | – | 49 ± 4 |
| Hydroxiapatite | 100 | – | – | – |
| Bending strength (MPa) | 50–150 a | 80–89 | 300 | 34 ± 2 |
| Elastic modulus (GPa) | 7–30 a | 105 | 170 | 77 ± 3 |
a The lowest values refer to trabecular (spongy) bone, the highest values to cortical (dense) bone [1] .
Fig. 6 shows characteristic fracture surfaces of strength specimens. No singular defects responsible for fracture were detected in any specimen. Fracture is tortuous with predominantly intergranular fracture.

Fig. 6. SEM images of the fracture surfaces of the sample sintered at 1250 °C/4 h; (a) general view, and (b) detail
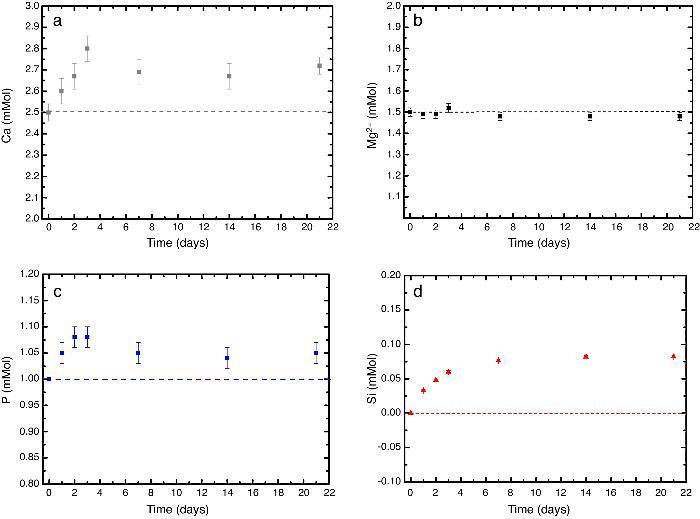
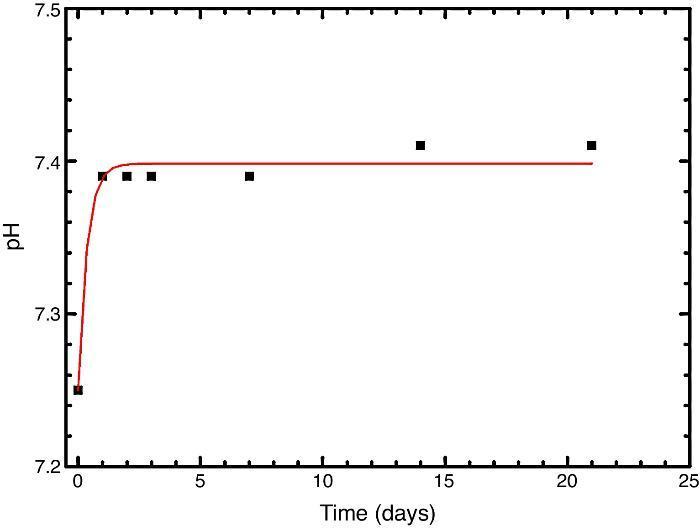
In vitro bioactivityIons release and pHFig. 7 shows the release profiles of Si, Ca, Mg and P ionic species in SBF for sintered specimens of the studied material. The concentration of Si, Ca and P ions increased sharply during the 4 first days of testing and then the concentration was stabilized (Si) or even decreased (Ca, P) during the remaining testing time. The Mg concentration did not change significantly along the experiment. The pH in the SBF bulk solution increased from a value of 7.25 ± 0.02 after 1 day and remained almost steady (∼7.4) during the rest of the monitoring period of 21 days (Fig. 8).
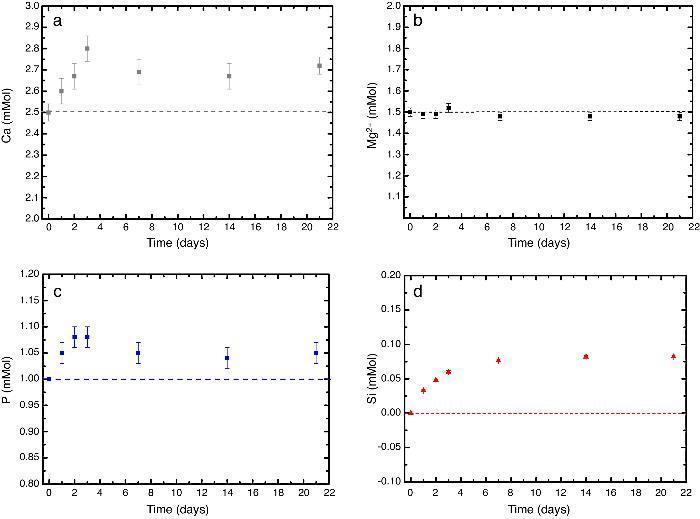
Fig. 7. (a–d) Variation of calcium, magnesium, phosphorus, and silicon ion concentration with immersion time in simulated body fluid at 37.0 ± 5 °C. Physiological values in human plasma are included as dashed line.
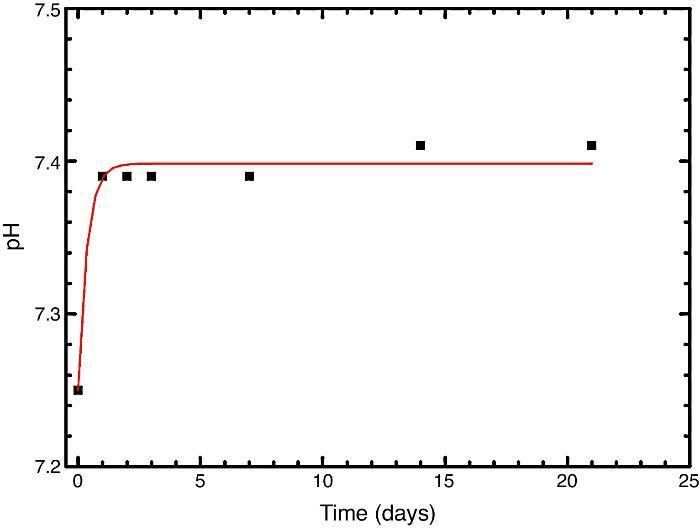
Fig. 8. Variation of pH with immersion time in simulated body fluid at 37.0 ± 5 °C. Physiological values in human plasma are included as dashed line.
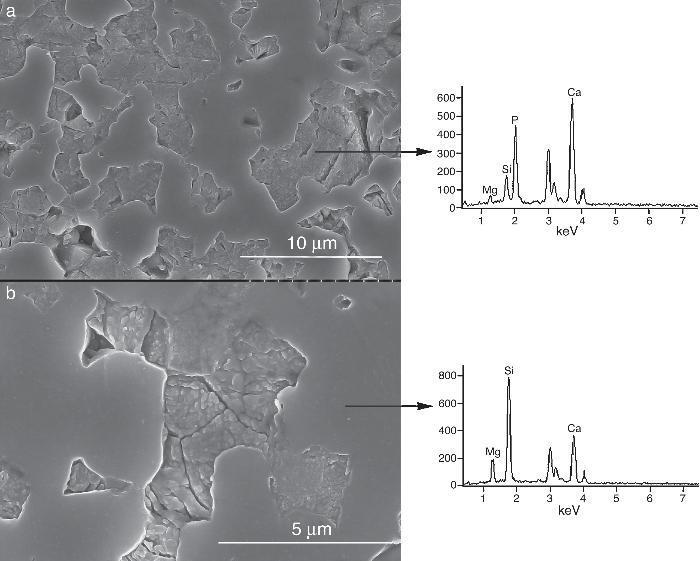
Fig. 9, Fig. 10 show the aspect of the surface of specimens before and after SBF testing and cross sections of tested specimens respectively. After experiment, surfaces were covered by a layer of globular particles of about 100 nm in diameter (Fig. 9 (b–d)), the layer covered the entire surface of the specimens after 3 weeks. The EDS analysis of the layers revealed Ca/P ratios of about 1.62.

Fig. 9. CFE-SEM micrographs of the ceramic surfaces after soaking in SBF, at 36.5 °C, for various times: (a) as sintering; (b) 7 days, (c) 14 days and (d) 21 days; SEM-EDS of Ap-like phase.
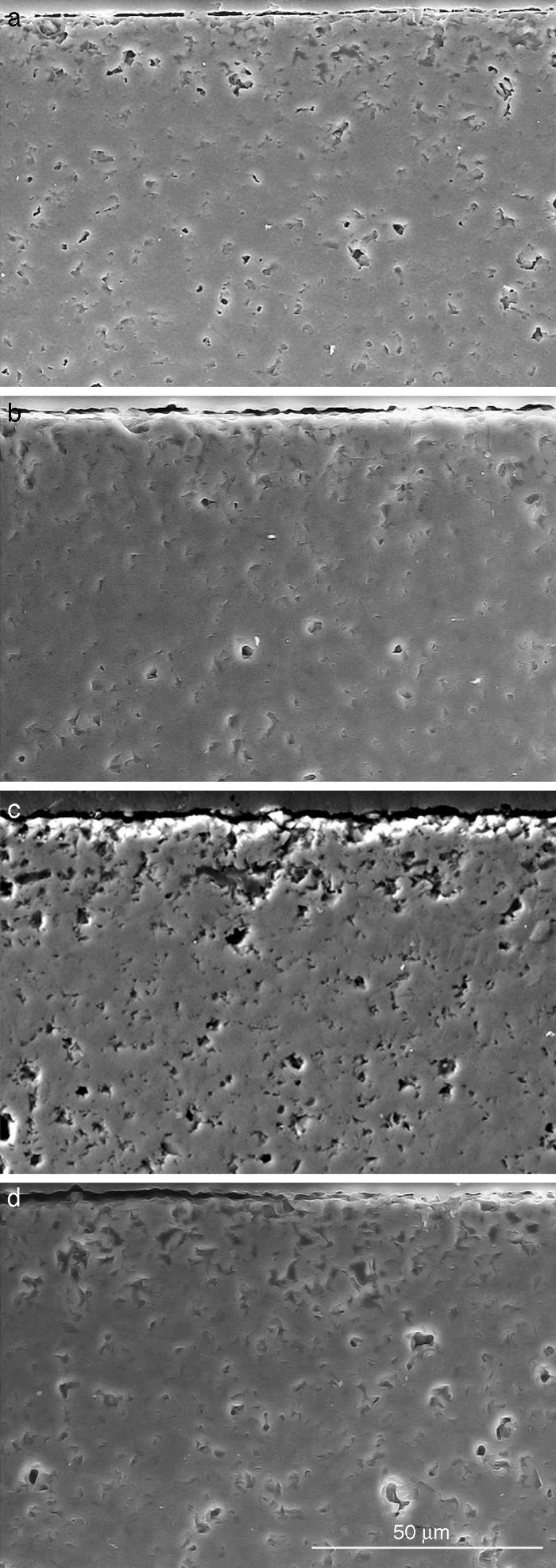
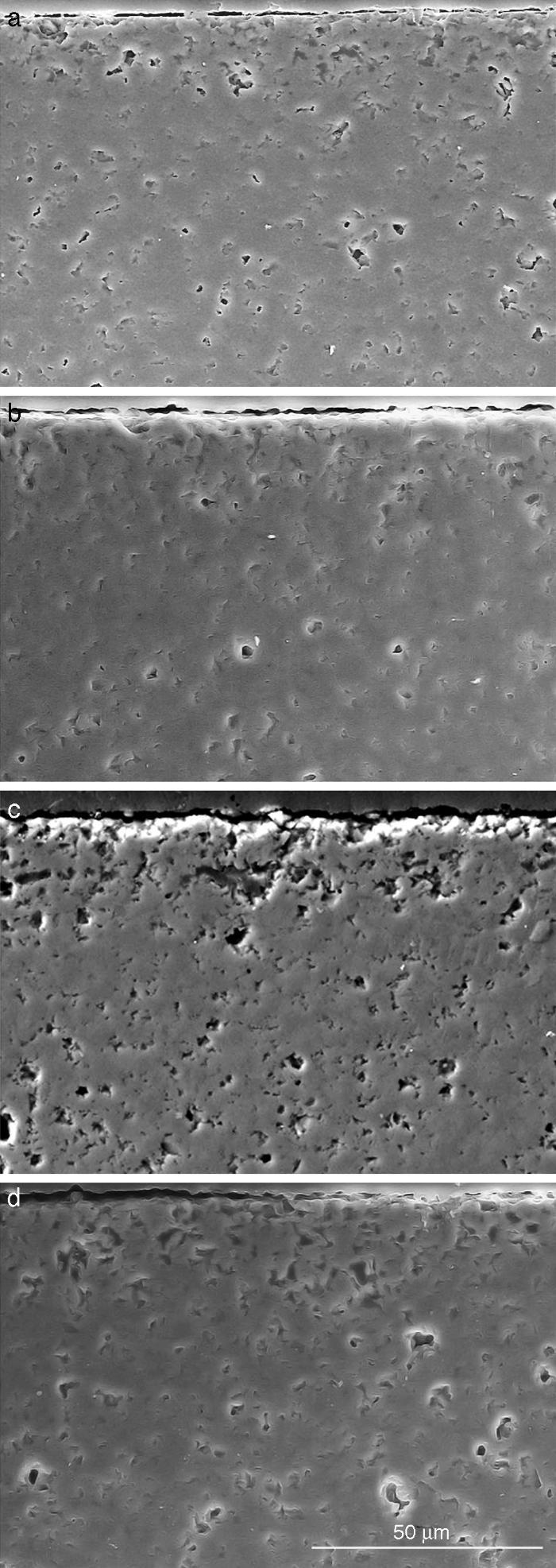
Fig. 10. Scanning electron microscopy micrographs of the cross sections of SBF/β-TCP-D sample (sintered at 1250 °C/4 h) interfaces after soaking for: (a) 3 days, (b) 7 days; (c) 14 days and (d) 21 days.
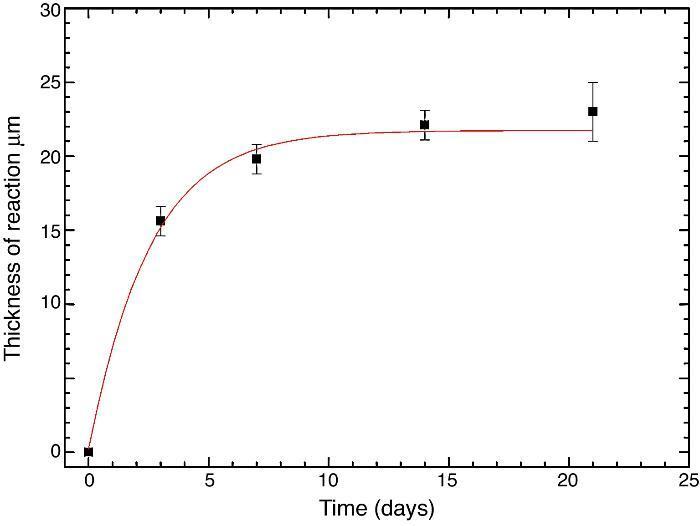
Fig. 10 reveals the presence of a porous layer between the original dense material and the globular particles. The thickness of the porous layer increased sharply (≈2.5 mm/day) during the first three days of soaking and then it increased slightly (≈0.8 mm/day) up to 7 days and remained practically constant during the remaining testing time (Fig. 11).
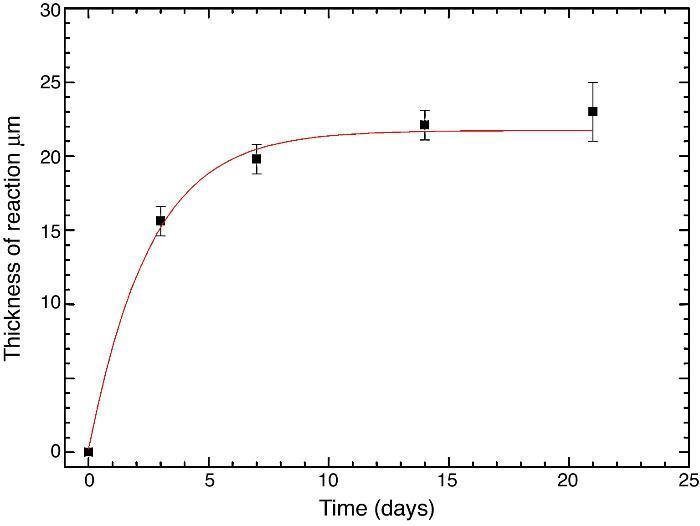
Fig. 11. Thickness change of the formed porous layer as a function of soaking time simulated body fluid.
Thermodynamic simulationA simulation of dissolution of β-Ca3(PO4)2, CaMg(SiO3)2 and a mixture of β-Ca3(PO4)2–CaMg(SiO3)2 (40:60 wt.%) was estimated by thermodynamic calculations using the HSC software ([22], [25], [26]). The composition of the original SBF was Na+ 142.0, K+ 5.0, Mg2+ 1.5, Ca2+ 2.5, Cl− 147.8, HCO3− 5.0, HPO42− 1.0 and SO42− 0.5 mol/m3 while HCl was added to fit the initial pH to 7.4. The species considered in the modelization of the aqueous phase were: H2O, Cl−, HCO3−, HPO42−, H2PO4−, PO43−, K+, Mg2+, MgOH+, Na+, OH−, H+, CaOH+, HSO4−, SO42−, HSiO3−, SiO44−. The temperature considered was 36.5 °C.
The total concentration of ions in solution was slightly greater than 0.3 mol/m3 being the major Na+ and Cl−. The solution was buffered to pH = 7.25–7.4 by a mathematical calculus.
In addition to the ionic species initially in the SBF, ions associated to the compounds that may precipitate during testing were included in the simulation. Furthermore, hydroxyapatite was considered as solid specie with the aim of studying the possibility of its precipitation from solution.
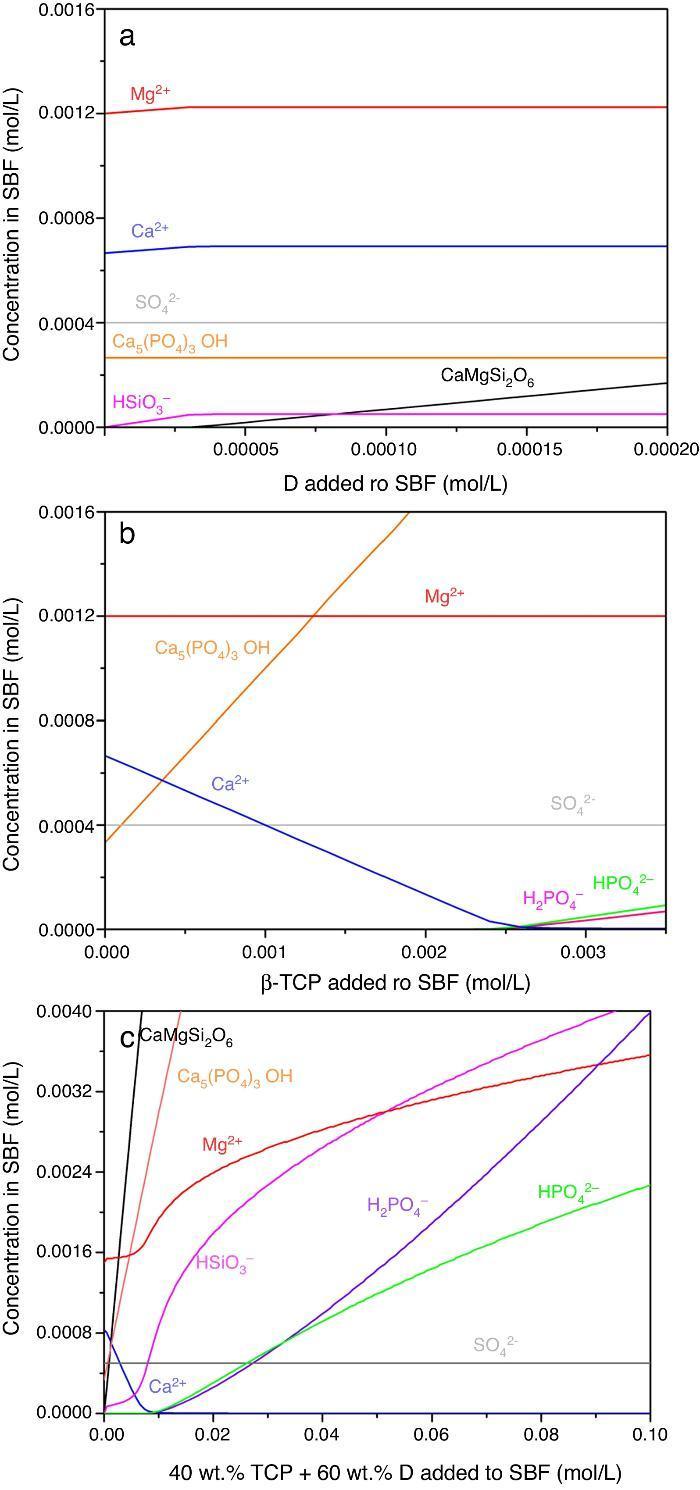
Result of the simulation of dissolutions of 1 g of Ca3(PO4)2, CaMg(SiO3)2 and mixtures β-Ca3(PO4)2–CaMg(SiO3)2 (40:60 wt.%) in 1 L of SBF are plotted in Fig. 12.
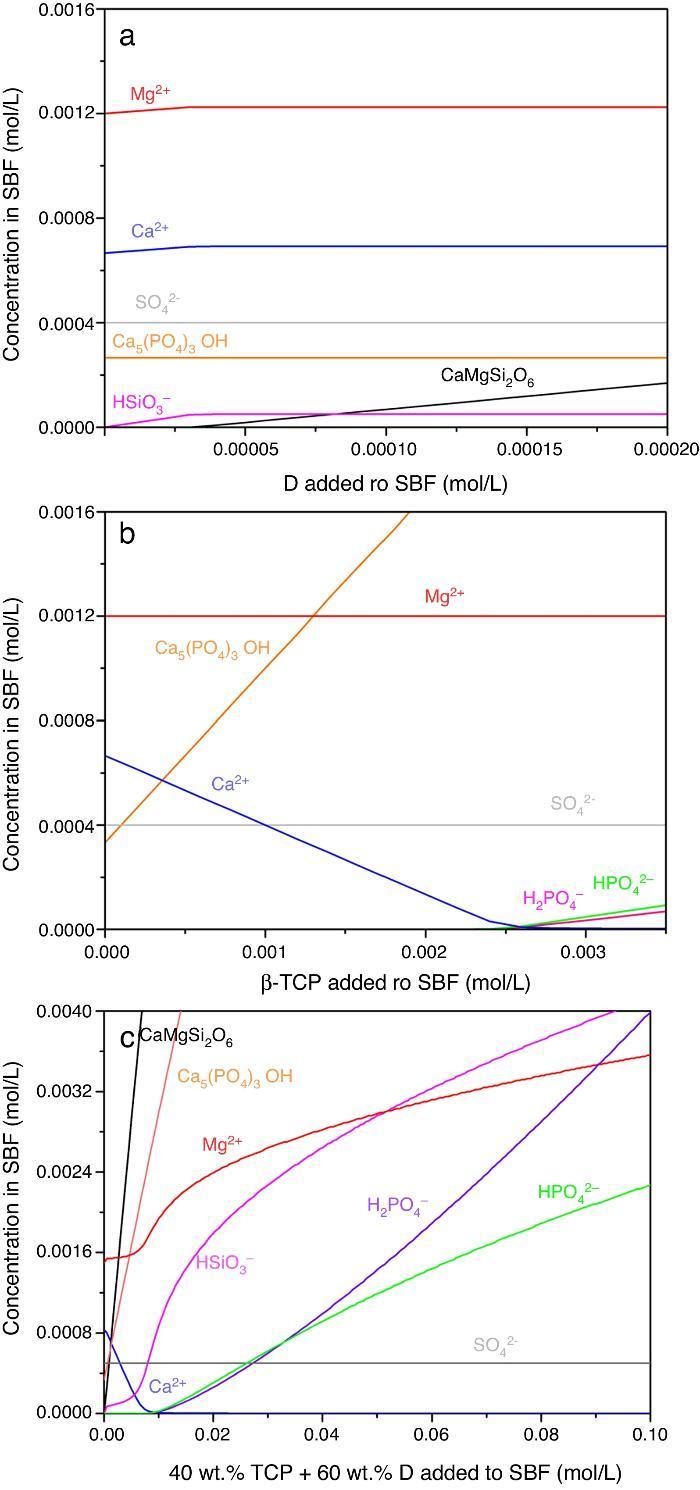
Fig. 12. Thermodynamic simulation of the dissolution behaviour of (a) diopside, (b) β-tricalcium phosphate and (c) mixture β-Ca3(PO4)2 – CaMg(SiO3)2 (40:60 wt.%) in 1 L of SBF at 37 °C. The composition of the aqueous phase has been represented as a function of the solid phase added.
Dissolution of CaMg(SiO3)2 in SBF up to about 3.14 × 10−5 mmol/L (Fig. 12a) is accompanied by an increase of the concentration of ions (HSiO3−, Mg2+, Ca2+). Once dissolution stops, the ion concentrations remain stable. A constant small amount of Ca5(PO4)3OH (≈0.0003 mmol/L) is expected in the SBF through the whole simulated range.
Dissolution of β-Ca3(PO4)2 in SBF (Fig. 12b) does not have a limit, as demonstrated by the lack of this compound in SBF through the whole simulated range. It is accompanied by the formation of significant amounts of Ca5(PO4)3OH. The amount of Ca2+ decreases and those of Mg2+ SO42+ remain constant while H2(PO4)− and H(PO4)2− are only observed for relatively large amounts (>0.0025 mmol/L) of β-Ca3(PO4)2 added.
In the Ca3(PO4)2–CaMg(SiO3)2 (40:60 wt.%) mixture, the maximum level of dissolution of diopside is higher (≈2×) than when simulation for the single compound is done (Fig. 12a) and β-Ca3(PO4)2 dissolves completely. Significant amounts of hydroxyapatite are formed through the whole simulated range, as occurred when β-Ca3(PO4)2 was considered as the single compound.
DiscussionMaterialsThe sintering behaviour of the green compacts, with a sharp shrinkage increase at temperatures around 1250 °C (Fig. 2a), can be explained by the presence of liquid. This temperature is slightly below the eutectic temperature of the pseudobinary system Ca3(PO4)2-CaMg(SiO3)2 (1300 °C, Fig. 1a). The presence of impurities (<1 wt.%, SiO2, MgO, Fe2O3 and Ca2P2O7) might locally decrease this temperature of first liquid formation in the compacts, leading to the formation of liquid phases ([15], [16], [27]). In fact, the density of the compacts sintered at 1250 °C was much higher than that of those sintered at 1225 °C, which can be explained by the presence of a small amount of liquid. The higher amount of liquid formed at a higher temperature in the compact sintered at 1275 °C led to swallowing of the material, as revealed by the large round pores observed (Fig. 4c).
The optimized composite, sintered at 1250 °C, showed a homogeneous microstructure consisting of β-Ca3(PO4)2 ss and CaMg(SiO3)2 areas. Those of β-Ca3(PO4)2 presented microcracks both at the boundaries with CaMg(SiO3)2 (Fig. 5a) and traversing them (Fig. 5b).
Considering the average thermal expansion coefficient of CaMg(SiO3)2, (α25–1000°C = 11.1 × 10−6 °C−1) and β-Ca3(PO4)2 (α500–1000°C = 20.2 × 10−6 °C−1), the formation of circumferential cracks at the boundaries can be attributed to the thermal expansion mismatch between these phases. Taking as model that of high thermal expansion TCP particles in a low thermal expansion D matrix, the β-Ca3(PO4)2 particles will contract more than the CaMg(SiO3)2 matrix during cooling from the sintering temperature, leading to stresses perpendicular to the particle-matrix boundaries. For particles size larger than the critical, cracks will be formed.
The formation of cracks traversing the β-Ca3(PO4)2 zones should be explained by the large crystalline thermal expansion anisotropy of this phase (α500–1000°C for a = 11.7 × 10−6 and b = 37.35 × 10−6 °C−1). Thermal expansion mismatch will arise at the boundaries of grains with different orientation and the developed stresses will also lead to microcracking. The microstructure of the obtained β-Ca3(PO4)2-CaMg(SiO3)2 composite explains the values of the Young's modulus and strength obtained, which were close to those of the cortical bone (Table 2) ([1]).
The Young's modulus of a composite can be estimated from those of its constituents, Ei, and their volume fractions, Vi, using the Voigth limit ([28]):
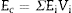
From the values of Table 2 for fully dense β-TCP and diopside and the volume fractions (0.46 and 0.54 for β-TCP and D respectively) a value of 137 GPa is obtained. Correcting this value for the porosity value (P ≈ 5%) using the well-known exponential relationship derived from the minimum solid area model:
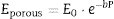
The features observed in the tortuous fracture surfaces (Fig. 6), in which no critical defects were observed, are responsible for the low strength values. Fracture will occur by the coalescence of microcracks present in the material and/or formed during loading due to thermal stresses. This kind of fracture leads to low and reproducible strength values ([29]), as those determined for the studied material.
In vitro bioactivityDuring the reaction of the ceramic composite in SBF it was found that calcium, phosphorus and silicon ion concentrations increased over the exposure time, indicating partial surface dissolution of the β-Ca3(PO4)2 and CaMg(SiO3)2 during the experiment. Diopside dissolution occurred in a much lesser extent (Fig. 7, silicon release).
The result of the SBF immersion was the formation of a porous surface structure constituted by the remainder diopside phase with small globules of Ca5(PO4)3OH inside the pores. As the dissolution progressed, the Ca2+, OH−, H2PO4− and HPO42− activities overcame the solubility product of apatite and this starts to precipitate with the typical morphology of globules constituted by aggregates of very fine acicular crystals (Fig. 9b and d). Although the formation of the Ca–P phase consumed some calcium ions, the calcium ion dissolution from the Ca3(PO4)2 were more than those consumed.
Taking into account the solubility simulations and the experimental results, the formation of the apatite layer on diopside-tricalcium phosphate ceramics can be summarized in the following steps:
Dissolution of β-Ca3(PO4)2
β-Ca3(PO4)2 + 3H+ → 3Ca2+ + H2PO4− + HPO42−
Dissolution of diopside
CaMgSi2O6 + 2H+ → 2HSiO3− + Ca2+ + Mg2+
Precipitation of hydroxyapatite
If considering the possibility of precipitation of apatite, the following reaction would occur:
5Ca2+ + 3 H2PO4− + H2O → Ca5(PO4)3OH + 7H+
Considering all possible reactions, dissolution of tricalcium phosphate and diopside other reaction of apatite precipitation would occur.
7CaMgSi2O6 + 2H2O + (3 − x) Ca2+ + (6 − y)H2PO4− → (Ca10 − x,Mgx)(PO4)6 − y(SiO4)y(OH)2 − y + (14 − y) HSiO3− + (7 − x)Mg2+
According to the simulation results the dissolution of diopside causes a decrease in the concentration of Ca2+ and HPO4− in SBF, whereas β-Ca3(PO4)2 dissolution causes an increase in the concentration of both ions in SBF. Therefore simultaneous dissolution in SBF of diopside and tricalcium phosphate produces a synergy and the solubility of both phases increases.
With the release of the silicon anions from the diopside many – (SiOH) groups are formed on the surfaces of β-Ca3(PO4)2-CaMg(SiO3)2 composites. These silanol groups induce heterogeneous nucleation of apatite, and the released calcium ions increase the ionic activity product of apatite enhancing apatite nucleation.
After 1 week, the surface was completely coated with nano-globules of crystal aggregates of calcium deficient hydroxyapatite (Ca/P = 1.62). Probably, SiOH groups existing on the remaining CaMg(SiO3)2 act as nucleating sites for apatite. New apatite layers are formed subsequently, with immersion time, coating the surface of the β-Ca3(PO4)2-CaMg(SiO3)2 ceramic.
The majority dissolution of β-Ca3(PO4)2 grains occurred according to Eq. (1), and Ca and P ions were released, increasing of Ca2+, OH−, HPO42− ionic activities at the neighbourhood the reacting surface until they overcome the solubility product of the hydroxyapatite. As β-Ca3(PO4)2 grains were dissolved, a porous surface layer, rich in diopside, was formed.
The results evidence that the microstructure morphology generated during immersion in the SBF was controlled by the solubility of the different phases, which generates changes of the surface chemistry and the surface topography. This mechanism leads to the in situ formation of an interconnected porous structure of diopside which could be expected to promote bone growth. This was confirmed for β-Ca3(PO4)2-CaMg(SiO4)2 materials by the results of the elemental EDS microanalysis performed on polished cross sections of these materials submitted to immersion in SBF for 3 weeks (Fig. 10). The phase with highest solubility, β-Ca3(PO4)2, controls the hydroxyapatite layer formation on the surface of diopside, which is the phase that produced the structural support of the biomaterial.
In previous reports ([10], [11], [12], [13]) it has been clearly established that synthetic diopside and tricalcium phosphate are not cytotoxic and are biocompatible, and osteogenic. Therefore the composite material presented here will be a promising resorbable biomaterial with controlled dissolution behaviour for temporary bone substitution. Additionally by means of processing science, it is possible to tailor Ca3(PO4)2-CaMg(SiO4)2 bioceramics with controlled microstructures in order to improve their osteointegration. The ionic dissolution products (Si, Mg and Ca ions) showed a range of concentration that will lead to enhanced proliferation of osteoblast (osteostimulation).
ConclusionsDense bioceramics based on tricalcium phosphate were successfully prepared by solid state sintering at 1250 °C for 4 h of homogeneous green bodies of tricalcium phosphate–diopside mixtures. The composite obtained showed a homogeneous fine grained microstructure constituted by two phases: one bioactive diopside (60 wt%, ∼6 μm) and the other resorbable β-CaMg(PO4)2 ss (40 wt.%, ∼3 μm). The thermal expansion mismatch between CaMg(SiO3)2 and β-Ca3(PO4)2 as well as the thermal expansion anisotropy of β-Ca3(PO4)2 led to microcracking of the material during cooling from the sintering temperature.
The presence of microcracks led to low values of strength (34 MPa) and elastic modulus (77 GPa), near to the ones of human bone. Tricalcium phosphate–diopside (40:60 wt.%) composite showed in vitro bioactivity. A globular apatite layer grew on its surface by immersion in simulated body fluid. Apatite formation occurs through a mechanism consisting on dissolution of β-Ca3(PO4)2, subsequent partial dissolution of CaMg(SiO3)2 and finally, nucleation and growth of an apatite layer similar to bone-like apatite on its porous surface. The in situ formation of an interconnected porous apatite-like/diopside layer at the ceramic-SBF interface was controlled by the enhanced solubility of β-Ca3(PO4)2 in presence of CaMg(SiO3)2 A simulation of dissolution behaviour of pure CaMg(SiO3)2 and Ca3(PO4)2 and of the 40:60 composite in SBF confirmed the established mechanism of bioactivity in this material.
Acknowledgements
This work was financially supported by the Spanish Government MINECO under project CICYT MAT2013-48426-C2-1-R and CSIC-201460E066. This work was made under the U.E. Cost Action MP1301 “Biomimetic and customized implants for bone engineering”. I. García-Páez also wishes to acknowledge financial support from Colciencias and the Latin American Scholarship Program of American Universities – Laspau.
Received 3 August 2015
Accepted 23 October 2015
Corresponding author. ppena@icv.csic.es

