




Q fever is a worldwide zoonosis caused by Coxiella burnetii—a small obligate intracellular Gram-negative bacterium found in a variety of animals. It is transmitted to humans by inhalation of contaminated aerosols from urine, feces, milk, amniotic fluid, placenta, abortion products, wool, and rarely by ingestion of raw milk from infected animals. Nested PCR can improve the sensitivity and specificity of testing while offering a suitable amplicon size for sequencing. Serial dilutions were performed tenfold to test the limit of detection, and the result was 10× detection of C. burnetti DNA with internal nested PCR primers relative to trans-PCR. Different biological samples were tested and identified only in nested PCR. This demonstrates the efficiency and effectiveness of the primers. Of the 19 samples, which amplify the partial sequence of C. burnetii, 12 were positive by conventional PCR and nested PCR. Seven samples—five spleen tissue samples from rodents and two tick samples—were only positive in nested PCR. With these new internal primers for trans-PCR, we demonstrate that our nested PCR assay for C. burnetii can achieve better results than conventional PCR.
Q fever is a worldwide zoonosis caused by Coxiella burnetii, a small obligate intracellular bacteria and Gram-negative pleomorphic member of the order Legionellales.1 It remains infectious in the environment for a long time and is resistant to chemical and physical factors.2 The infection exists in a variety of animals including various ruminants, domestic and wild animals, birds, and arthropods (especially ticks).2 Cattle, sheep, and goats are the primary reservoirs of C. burnetii.2–5 Transmission to humans usually occurs through inhalation of contaminated aerosols from urine, feces, milk, amniotic fluid, placenta, abortion products, wool, or less commonly through ingestion of raw milk from infected animals.2,5
There are various ways of detecting C. burnetii infection.3 Several PCR-based DNA detection methods have been used to diagnosis Q fever including conventional PCR, nested PCR, real-time PCR (qPCR), and the loop-mediated isothermal amplification (LAMP) assay.3,6,7 Berri et al.8 showed that the sensitivity of trans-PCR is 100-fold greater than that obtained by PCR using primer CB1–CB2. Complete sequencing of the C. burnetii genome showed 20 copies of the IS1111 transposase. There were many element number copies in the genome of C. burnetii, and this was a useful tool for the detection and differentiation of C. burnetii isolates.9 The objective of this study was to increase the sensitivity of the trans-PCR primer using a nested PCR assay and to demonstrate its effectiveness in different species of animals and different types of samples.
Materials and methodsClinical samples and DNA extractionPatient samples (n=24) with suspected Q fever were tested along with 68 domestic animals, 74 rodents, and 180 ticks from Rio de Janeiro, Brazil. Domestic animal samples and ticks were obtained from areas where C. burnetii infection was detected in human and animals,10–12 and rodent samples were collected from various regions of Rio de Janeiro state as part of an ecological research project conducted on wild animals from 2008 and 2015. Conventional and nested PCR were used to assay biological specimens from the suspected Q fever cases as well as clot, tissue, milk, and anal and vaginal swab samples extracted from animals. In addition, tissue samples from rodents and ticks were also tested with conventional and nested PCR.
DNA was extracted from goats, sheep, rodents, dogs, and cats. Serum and clot samples were used for human DNA extraction. The samples were tested using a QIAamp DNA Mini Kit (Qiagen™, Valencia, CA, USA) according to the instruction manual. After extensive washing in PBS, the DNA from milk as well as anal and vaginal swabs was extracted using ATL (QIAamp DNA Mini Kit, Qiagen™) and proteinase K. Each sample (200μL) was mixed directly with the ATL and proteinase K and incubated at 56°C overnight. This was followed by an incubation step with AL buffer for 10min at 70°C.10,13 The extraction process continued with a QIAmp DNA kit (QIAamp DNA Mini Kit, Qiagen™) following the manufacturer's instructions. Negative controls were included in each extraction to check for possible DNA contamination.
Obtaining and purifying C. burnetii DNAC. burnetii Nine Mile phase II strain isolate from the UNIFESP São Paulo Culture Collection was used as a the positive control.14 According to Zamboni and collaborators, the stock suspensions containing the bacteria were mildly sonicated at 35kHz for 15min at room temperature to disrupt aggregates. C. burnetii were grown in Vero cells (about 3×105 cells per 2cm2). In 2–3 days, the Vero cells developed large vacuoles containing bacteria. There were 3–10μL of bacterial stock per well. After 24h, cultures were vigorously washed and fresh medium added (minimal essential medium, MEM, containing 15mM 4-(2-hydroxy-ethyl)-1-piperazineethanesulfonic acid – HEPES, 2g/L sodium bicarbonate, 1mM l-glutamine and 5% (v/v) of fetal bovine serum).14 The DNA was extracted from 200μL of this solution using a QIAamp DNA Mini Kit (Qiagen™, Valencia, CA, USA) following the manufacturer's instructions. The DNA was then quantified and used as a positive control for the first PCR. Serial tenfold dilutions were used to examine the detection limit.
PCR assayThe PCR assay used a pair of primers that target the gene IS1111 transposase elements in the genome of C. burnetii.15,16 The expected amplification product using these primers was 687bp. The pair of primers were Trans1 (5′TAT GTA TCC ACC GTA GCC AGT C-3′) and Trans2 (5′-CCC AAC AAC ACC TCC TTATTC-3′). These primers were used to do a PCR reaction using 4μL of each DNA sample in 25μL final reaction volume. The final reaction mixture contained 1× PCR buffer 10×, 0.2μM of each primer (RTD/Prodimol™, Belo Horizonte, MG, Brazil), 1.5mM of MgCl2, 200μM of dNTP mix (20mM of each deoxynucleotide triphosphate), 0.5U of Platinum Taq DNA polymerase (Invitrogen™, Carlsbad, CA, USA), 4μL of DNA sample, and nuclease-free water (Promega™, Madison, WI, USA). Amplification used a GeneAmp PCR System 9700 thermal cycler (Applied Biosystems™, Foster City, CA, USA): initial denaturation at 95°C for 5min, followed by 40 successive cycles of denaturation at 95°C for 30s, annealing at 60°C for 30s, and extension at 72°C for 1min. This was followed by a final extension of 7min at 72°C. All steps used negative controls to control contamination.
Nested PCR assayPrimers designOligonucleotide primers to detect C. burnetii were designed from conserved regions of different IS1111 gene sequences. The MUSCLE tool was used to perform multiple sequence alignment17; MUSCLE is part of the MEGA 6 software package.18 The nucleotide sequences of the IS1111 gene were analyzed (GenBank accession no. AE016828, CP001020, CP001019, CP000890, CP000733, CP007555, HG825990, LK937696, and M80806). The highly conserved regions were identified, and the degenerate oligonucleotide primers were designed to correspond to nucleotides in these regions.
Standardization and gradientWe used primers from the trans-PCR for the first PCR; N3+ (5′-AAG CGT GTG GAG GAG CGA ACC-3′) and N4− (5′-CTC GTA ATC ACC AAT CGC TTC GTC-3′) were used for the second PCR designed from the gene IS1111 transposase elements in the genome of C. burnetii.15,16 The expected amplification product of the target sequence with these primers was 440bp long. The 25μL mixture contained 2μL of DNA samples (2μL of first PCR reaction mixture in second PCR). The final reaction mixture contained 1× PCR buffer 10×, 0.2μM of each primer (RTD/Prodimol™, Belo Horizonte, MG, Brazil), 1.5mM of MgCl2, 200μM of dNTP mix (20mM of each deoxynucleotide triphosphate), and 0.5U of Platinum Taq DNA polymerase (Invitrogen™, Carlsbad, CA, USA).
A gradient PCR amplification was performed in a Veriti 96-well thermal cycler (Applied Biosystems™, Foster City, CA, USA). This consisted of initial denaturation for 5min at 95°C, followed by 30 successive cycles of denaturation at 95°C for 30s, annealing in a gradient of 65/66/67/68/69/70°C for 30s, and extension at 72°C for 1min. This was followed by a final extension of 5min at 72°C. To confirm amplification, the products were analyzed by electrophoresis in 1% agarose gel stained with GelRed™ solution (Biotium, Hayward, CA, USA). A 100-bp DNA ladder (Invitrogen™, Carlsbad, CA, USA) served as a molecular weight marker. The amplified products were visualized under a UV light trans-illuminator and stored in a digital system for gel documentation (Carestream Gel Logic 212 Pro, Rochester, NY, USA) to select the annealing temperature. Thus, the selected reaction used initial denaturation for 5min at 95°C followed by 30 successive cycles of denaturation at 95°C for 30s, annealing at 66°C for 30s, extension at 72°C for 30s, and a final extension to 72°C for 5min.
Sensitivity of PCR and nested PCR assaysVero cells with C. burnetii were extracted using the QIAamp DNA Mini Kit (Qiagen™, Valencia, CA, USA) following the manufacturer's instructions. After extraction, the total DNA was quantified using a Qubit© 2.0 fluorometer (Invitrogen™, Carlsbad, CA, USA) indicating a concentration of 21.4μg/mL. Solutions of purified C. burnetii were used as positive control, and dilutions were prepared ranging from 1 to 10.13 Dilutions used 2μL of the solution extracted with 18μL of nuclease-free water until the 1013 dilution. Four μL of the DNA solution was used for the PCR, and 2μL was used for the nested PCR assays.
Specificity of PCR and nested PCR assaysDNA samples of C. burnetii and 15 other bacteria were used in the PCR and the nested PCR assays to evaluate specificity. The bacteria were Rickettsia rickettsii, Rickettsia typhi, Ehrlichia chaffeensis, Ehrlichia canis, Anaplasma phagocytophilum, Bartonella henselae, Borrelia burgdorferi (Hantaviroses and Rickettsioses Laboratory, FIOCRUZ), Pseudomonas aeruginosa INCQS 00025 (ATCC 15442), Brucella abortus INCQS 00242 (ATCC 7705), Legionella pneumophila subsp. pneumophila INCQS 00451 (NCTC 11232 ATCC 33155), Staphylococcus aureus CCBH 3853 (ATCC 25923), Streptococcus pneumonia, Haemophilus influenzae, Neisseria meningitidis (Epidemiology and Molecular Systematics Laboratory, FIOCRUZ), and Listeria monocytogenes (Bacterial Zoonoses Laboratory, FIOCRUZ).
ResultsSensitivity of the PCR and nested PCR assaysSensitivity was compared between the PCR and the nested PCR assays in the detection of C. burnetii DNA. The amplification product by PCR assay with the trans-PCR primers for the first PCR was detected until the 108 dilution (Fig. 1). The first PCR data showed that the annealing temperature gradients were negative for solutions of 109 to 1013. The detection by nested PCR assay with primers N3+ and N4− was effective to the 109 dilution indicating that it is 10-fold more sensitive than the first PCR assay (Fig. 2). Thus, the selected reaction was the initial denaturation for 5min at 95°C followed by 30 successive cycles of denaturation at 95°C for 30s, annealing at 66°C for 30s, extension at 72°C for 30s, and a final extension to 72°C for 5min.
![PCR assay with C. burnetii performed with primers of first PCR as described in the materials and methods. Lanes: 1, negative control; 2, 100bp DNA ladder; 3, C. burnetii suspension 1× [21.4μg/mL]; 4, C. burnetii suspension 10×; 5, C. burnetii suspension 102×; 6, C. burnetii suspension 103×; 7, C. burnetii suspension 104×; 8, C. burnetii suspension 105×; 9, C. burnetii suspension 106×; 10, C. burnetii suspension 107×; 11, C. burnetii suspension 108×; 12, C. burnetii suspension 109×; 13, C. burnetii suspension 1010×; 14, C. burnetii suspension 1011×; 15, C. burnetii suspension 1012×; and 16, C. burnetii suspension 1013×.](https://static.elsevier.es/multimedia/15178382/0000004900000001/v2_202006110912/S1517838216304014/v2_202006110912/en/main.assets/gr1.jpeg?xkr=ue/ImdikoIMrsJoerZ+w997EogCnBdOOD93cPFbanNerEcKXq/4gYMpNkXqSlVDY9W8lBgxJd7sP21C796+Vu43H9IIFkf/r+xpkAZ+WPeUdpGUAlGY0jWV7f3Awc0QE1Nkx3zK+pT1Y02KOLXWWQGuZHkvWt+bFsB77iBgpdty3HUnmTWrBz5E2LTxZZ07udIIfTacpUY9WcKTs8l+yMqalMppkDAPkT7gxGhctKb/D0KB1JZ4Jd8QGgRCbuhyuLcnDzXJJnr7avoPiVMmnxnTh7MW8IwozRHNG0Mk3ca0= "PCR assay with C. burnetii performed with primers of first PCR as described in the materials and methods. Lanes: 1, negative control; 2, 100bp DNA ladder; 3, C. burnetii suspension 1× [21.4μg/mL]; 4, C. burnetii suspension 10×; 5, C. burnetii suspension 102×; 6, C. burnetii suspension 103×; 7, C. burnetii suspension 104×; 8, C. burnetii suspension 105×; 9, C. burnetii suspension 106×; 10, C. burnetii suspension 107×; 11, C. burnetii suspension 108×; 12, C. burnetii suspension 109×; 13, C. burnetii suspension 1010×; 14, C. burnetii suspension 1011×; 15, C. burnetii suspension 1012×; and 16, C. burnetii suspension 1013×.")
PCR assay with C. burnetii performed with primers of first PCR as described in the materials and methods. Lanes: 1, negative control; 2, 100bp DNA ladder; 3, C. burnetii suspension 1× [21.4μg/mL]; 4, C. burnetii suspension 10×; 5, C. burnetii suspension 102×; 6, C. burnetii suspension 103×; 7, C. burnetii suspension 104×; 8, C. burnetii suspension 105×; 9, C. burnetii suspension 106×; 10, C. burnetii suspension 107×; 11, C. burnetii suspension 108×; 12, C. burnetii suspension 109×; 13, C. burnetii suspension 1010×; 14, C. burnetii suspension 1011×; 15, C. burnetii suspension 1012×; and 16, C. burnetii suspension 1013×.

PCR assay with C. burnetii performed with primers of nested PCR as described in the materials and methods. Lanes: 1, negative control of first PCR; 2, negative control of nested PCR; 3, 100bp DNA ladder; 4, C. burnetii suspension 1×; 5, C. burnetii suspension 10×; 6, C. burnetii suspension 102×; 7, C. burnetii suspension 103×; 8, C. burnetii suspension 104×; 9, C. burnetii suspension 105×; 10, C. burnetii suspension 106×; 11, C. burnetii suspension 107×; 12, C. burnetii suspension 108×; 13, C. burnetii suspension 109×; 14, C. burnetii suspension 1010×; 15, C. burnetii suspension 1011×; 16, C. burnetii suspension 1012×; and 17, C. burnetii suspension 1013×.
DNA samples from the 15 other bacteria used in the first PCR and nested PCR assays to assess the specificity showed no amplification (data not shown).
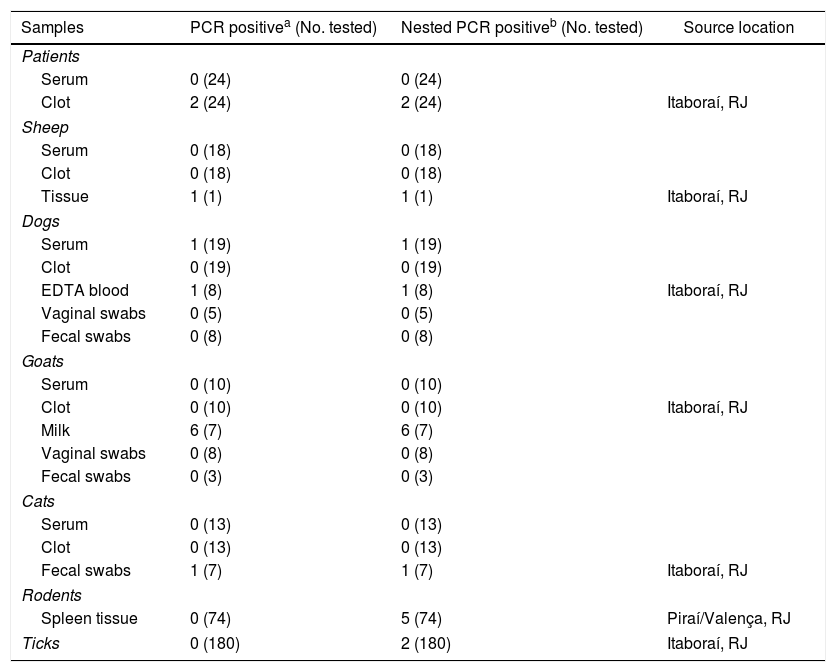
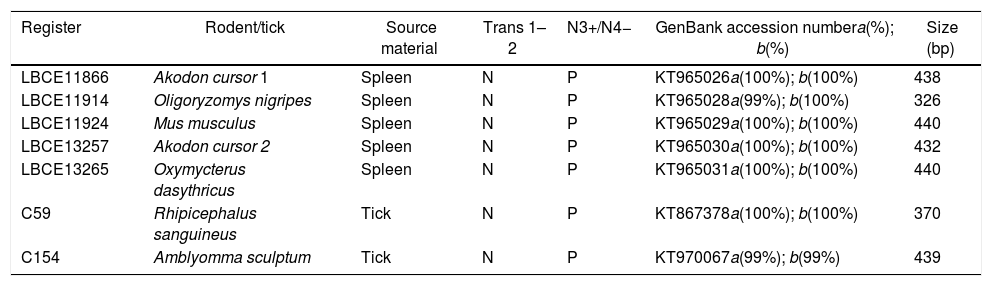
Detection of C. burnetii DNA from clinical specimensThe samples were tested for C. burnetii DNA by the first PCR and the nested PCR assays. Of 24 patients, 2 were positive (clot sample). One sheep (tissue) and two dogs (blood samples) were also positive. Of 10 goats, six were positive (milk sample), and of 13 cats, one was positive (anal swab) (Table 1). Of 74 rodents, the spleen tissue sample from five rodents and two tick samples of the 180 tested (Table 2) were only positive with the nested PCR. Therefore, they were sequenced, and the partial sequences were deposited in GenBank.
Detection of Coxiella infection by PCR of primers (trans 1/2) and nested PCR (+N3/N4) among human, domestic animals, rodents, and tick samples collected in Rio de Janeiro between 2008 and 2015.
| Samples | PCR positivea (No. tested) | Nested PCR positiveb (No. tested) | Source location |
|---|---|---|---|
| Patients | |||
| Serum | 0 (24) | 0 (24) | |
| Clot | 2 (24) | 2 (24) | Itaboraí, RJ |
| Sheep | |||
| Serum | 0 (18) | 0 (18) | |
| Clot | 0 (18) | 0 (18) | |
| Tissue | 1 (1) | 1 (1) | Itaboraí, RJ |
| Dogs | |||
| Serum | 1 (19) | 1 (19) | |
| Clot | 0 (19) | 0 (19) | |
| EDTA blood | 1 (8) | 1 (8) | Itaboraí, RJ |
| Vaginal swabs | 0 (5) | 0 (5) | |
| Fecal swabs | 0 (8) | 0 (8) | |
| Goats | |||
| Serum | 0 (10) | 0 (10) | |
| Clot | 0 (10) | 0 (10) | Itaboraí, RJ |
| Milk | 6 (7) | 6 (7) | |
| Vaginal swabs | 0 (8) | 0 (8) | |
| Fecal swabs | 0 (3) | 0 (3) | |
| Cats | |||
| Serum | 0 (13) | 0 (13) | |
| Clot | 0 (13) | 0 (13) | |
| Fecal swabs | 1 (7) | 1 (7) | Itaboraí, RJ |
| Rodents | |||
| Spleen tissue | 0 (74) | 5 (74) | Piraí/Valença, RJ |
| Ticks | 0 (180) | 2 (180) | Itaboraí, RJ |
Partial sequences of the gene IS1111 transposase elements in the genome of C. burnetii at GenBank from samples obtained from nested PCR.
| Register | Rodent/tick | Source material | Trans 1–2 | N3+/N4− | GenBank accession numbera(%); b(%) | Size (bp) |
|---|---|---|---|---|---|---|
| LBCE11866 | Akodon cursor 1 | Spleen | N | P | KT965026a(100%); b(100%) | 438 |
| LBCE11914 | Oligoryzomys nigripes | Spleen | N | P | KT965028a(99%); b(100%) | 326 |
| LBCE11924 | Mus musculus | Spleen | N | P | KT965029a(100%); b(100%) | 440 |
| LBCE13257 | Akodon cursor 2 | Spleen | N | P | KT965030a(100%); b(100%) | 432 |
| LBCE13265 | Oxymycterus dasythricus | Spleen | N | P | KT965031a(100%); b(100%) | 440 |
| C59 | Rhipicephalus sanguineus | Tick | N | P | KT867378a(100%); b(100%) | 370 |
| C154 | Amblyomma sculptum | Tick | N | P | KT970067a(99%); b(99%) | 439 |
a(%) maximum similarity with the sequence of the IS1111 gene of C. burnetii RSA 331.
b(%) query cover with the sequence of the IS1111 gene of C. burnetii RSA 331.
N, negative; P, positive.
Other samples including milk, vaginal swab, tissue, and serum samples had initial PCRs that were positive. They were then sequenced using the nested PCR. The tissue sample from sheep (GenBank accession no. KC854154 – 373bp), EDTA blood sample from dog (GenBank accession no. KT867377 – 439bp), and fecal swab from cat (GenBank accession no. KC854155 – 394bp) were also deposited in GenBank.
DiscussionPCR has been used widely for the diagnosis of infectious diseases including those caused by C. burnetti. Hemi-nested and nested PCR increase the sensitivity of PCR and lead to at least a 10,000-fold enhancement in PCR product over nonspecific products that could be co-amplified when using only the outer primers. The amount of PCR product used in the nested PCR should be adjusted according to the results of the first PCR. In future diagnostic work, all samples should be tested with nested primers regardless of the first PCR result. False negative results after the first PCR could occur due to a very small number of copies.19,20
The objective of this study was to design and use nested PCR for a target gene that has already been shown to have sufficient specificity and sensitivity.8,15,16 These new internal primers for trans-PCR proved that there was a 10× increase in sensitivity with an optimal sequence size of 440bp.
Although Boden and colleagues developed Nested PCR using the IS1111 element as the target,21 our study verified not only the presence of the partial sequence of the C. burnetii gene in serum samples but also used it to test for other samples used for epidemiological studies such as swabs, milk, and ticks. Samples for epidemiological research may have small amounts of microorganisms.22 In contrast to Duron,23 small size amplifications can actually characterize the detection of fragments and not C. burnetii in its whole form. Thus, we developed this primer, which is located at a different site and with a reasonable size for sequencing. We determined that our amplicons were different from the qPCR DNA sequences studied previously.24 The primers developed in the present study are not likely to result in amplification of endosymbionts due to the degradation in this particular region of their IS1111 gene.24
Pearson and colleagues24 detected Coxiella-like bacteria through the IS1111 target. These are rare in tick populations, which limits the impact of false positives on prevalence estimates of C. burnetii. We distinguished C. burnetii from Coxiella-like bacteria using C. burnetii SNP genotyping assays due to the extreme sensitivity of the IS1111 multiple copy assay.
Although several qPCR techniques have been shown to be sensitive for diagnosis,3,7 this study aimed to find an alternative to conventional PCR in laboratories with few applicable financial resources for the realization of a qPCR. Chen et al. also proposed a novel detection method for serum samples.6 However, nested PCR can be used in different types of samples such as milk, swabs, and tissue samples.25
Recently, the presence of IS1111 in both C. burnetii and in the Coxiella-like endosymbionts led to questioning the specificity of the diagnostic tests in Q fever epidemiology. However, IS1111 amino-acid sequences in Coxiella-like endosymbionts revealed the presence of degraded sequences containing an internal stop codon(s). These are likely non-functional.23 Thus, PCR or qPCR techniques for Q fever diagnosis using small fragments of the IS1111-DNA may lead to misidentification.21 Conversely, our new internal primers (N4−) for IS were developed considering the degraded regions for Coxiella-like endosymbionts. Therefore, we provide a nested PCR technique with better sensitivity for C. burnetii and a suitable DNA fragment size for genetic characterization.
Using these primers, we amplified and sequenced 440bp partial sequences that do not align with the 15 other bacteria tested here. This makes the primers a great alternative for qPCR. We also used samples from different hosts and different types, and we improved the specificity of the amplified material. In animals, we identified positive cases in sample types other than blood. This makes this technique valuable for surveillance. We demonstrated that our nested PCR assay for C. burnetii achieved better results than conventional PCR and that this method may be a good alternative for the diagnosis of C. burnetii infection.
Financial supportThis study was supported by FIOCRUZ and grants from Conselho Nacional para o Desenvolvimento Cientifico e Tecnológico (CNPq) Project 407664/2012 and Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Project E-26/010.001567/2014.
Conflicts of interestThe authors have no conflict of interest or disclosures concerning this paper.
Phase II Nine Mile strain of C. burnetii was provided by Dr. Fernando Real and Dr. Alexis Melo from EPM, UNIFESP, São Paulo, SP, Brazil. Bacterial DNA was kindly provided by Dr. Ivano de Filippis, Collection Reference Microorganisms in Health Surveillance, CMRVS Fundação Oswaldo Cruz – FIOCRUZ, Brazil; the Epidemiology and Molecular Systematics Laboratory – Fundação Oswaldo Cruz – FIOCRUZ, Brazil; and Dra. Martha Pereira, Bacterial Zoonoses Laboratory – Fundação Oswaldo Cruz – FIOCRUZ, Brazil.