


We examined microbial communities from enriched fine and retorted shale particles using sequencing of V4 variable region of 16S rRNA. High number of microbial genera was found in both enriched shale by-products that were dominate by Actinobacteria, Firmicutes and Proteobacteria, showing differences due to microbial colonization after the pyrolysis process.
Oil shale is defined as fine-grained sedimentary rock containing high amounts of organic matter that is predominantly kerogen, which will yield oil and combustible gas upon destructive distillation.1 Oil shale resources are abundant and distributed in several countries around the world. The most important Brazilian oil shale reserve, the Permian Irati Formation located in São Mateus do Sul, Paraná, has the greatest potential for economic development because of its accessibility, grade, and wide-spread distribution.1 The mining and processing of shale generate solid by-products that occur in two distinct stages the fine shale particles (FS) and retorted shale (RS). The FS are small fragments of shale rock measuring less than 0.25in. with an oil content of approximately 15% and are 20% of mined rock; thus, FS are not usable in oil and gas processing.2 Retorted shale (RS) is a waste by-product obtained from pyrolysis (550°C) of oil shale processing.3
Although the interaction of oil shale with the microbial community is an interesting topic, until now, this subject has not been well examined. Due to the lack of information regarding the occurrence of microorganisms in the shale by-products from the Irati Formation, knowledge of the microbial community composition can generate important data and suggests how bacteria can act in providing complex nutrients, thus adding value to by-products as agricultural inputs. In this perspective, the purpose of this study was to examine the composition of microbial communities from enriched FS and RS using sequencing of V4 variable region of the 16S rRNA gene.
Aiming to evaluate if there would be differences in the bacterial community over time sampling was carried out in 2008 (FSI and RSI) and 2011 (FSII and RSII) at Schist Industrialization Business Unit (Six) of Petrobras, in São Mateus, Paraná, Brazil (25°52′26″ S, 50° 22′58″ W) in storage piles of shale by-products as described by Goes et al.4 An aliquot of 10g from four samples of shale by-product were enriched in Luria Bertani (LB),5 J. E. medium6 and dispersed sterile water for 30 days prior DNA extraction using the MoBio Powersoil DNA isolation kit (Geneworks, Australia). Extracting high-purity DNA directly from environmental samples is essential for the study of microbial community composition. Direct DNA extraction was performed from by-products at least three times without success. To allow the proliferation of microorganisms that could grow on the medium provided and then make available extractable DNA; we used an enrichment method.7 The total amount of DNA extracted was quantified and checked for purity with a Qubit™ Fluorometer using Quant-iT™ dsDNA BR assay kit (Invitrogen™). Bacterial DNA was amplified using primers F515 (5′-GTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACHVGGGTWTCTAAT-3′) targeting the V4 region of the SSU rRNA gene.8 Barcoded libraries were sequenced on Ion 314™ Chip (10 Mb.p.) using the Ion Torrent Personal Genome Machine – PGM (Life Technologies, USA). Reads were filtered within the Ion Torrent server to remove low quality and polyclonal sequences. The resulting sequencing data sets were uploaded to the Metagenome Rapid Annotation using Subsystem Technology (MG-RAST) (http://metagenomics.nmpdr.org/), checked for low-quality reads prior to dereplication, assigned phylogenetic identification of phylum level using the Ribosomal Database Project (RDP) with 95% identity and the alpha diversity index calculation (http://rdp.cme.msu.edu/index.jsp).9
Differences between the taxonomical distribution were analyzed in the Statistical Analysis of Metagenomic Profiles program (STAMP).10 The data corresponding to phylum distribution were submitted to ANOVA statistical and a multiple groups comparison test by Tukey-Kramer test (p≤0.05) to construct a principal component analysis (PCA) plot and a similarity dendrogram (heatmap) using the UPGMA algorithm (Unweight Pair Group Method with Arithmetic mean). The mean proportion between the phylum level samples was analyzed by Fisher's statistical test, with Newcombe-Wilson confidence intervals (p≤0.05).
The two samples of RS and two of FS were enriched with two culture media, LB and J. E. media, with the intent of favoring different bacterial groups, however only LB medium allows DNA extraction and PCR amplification.
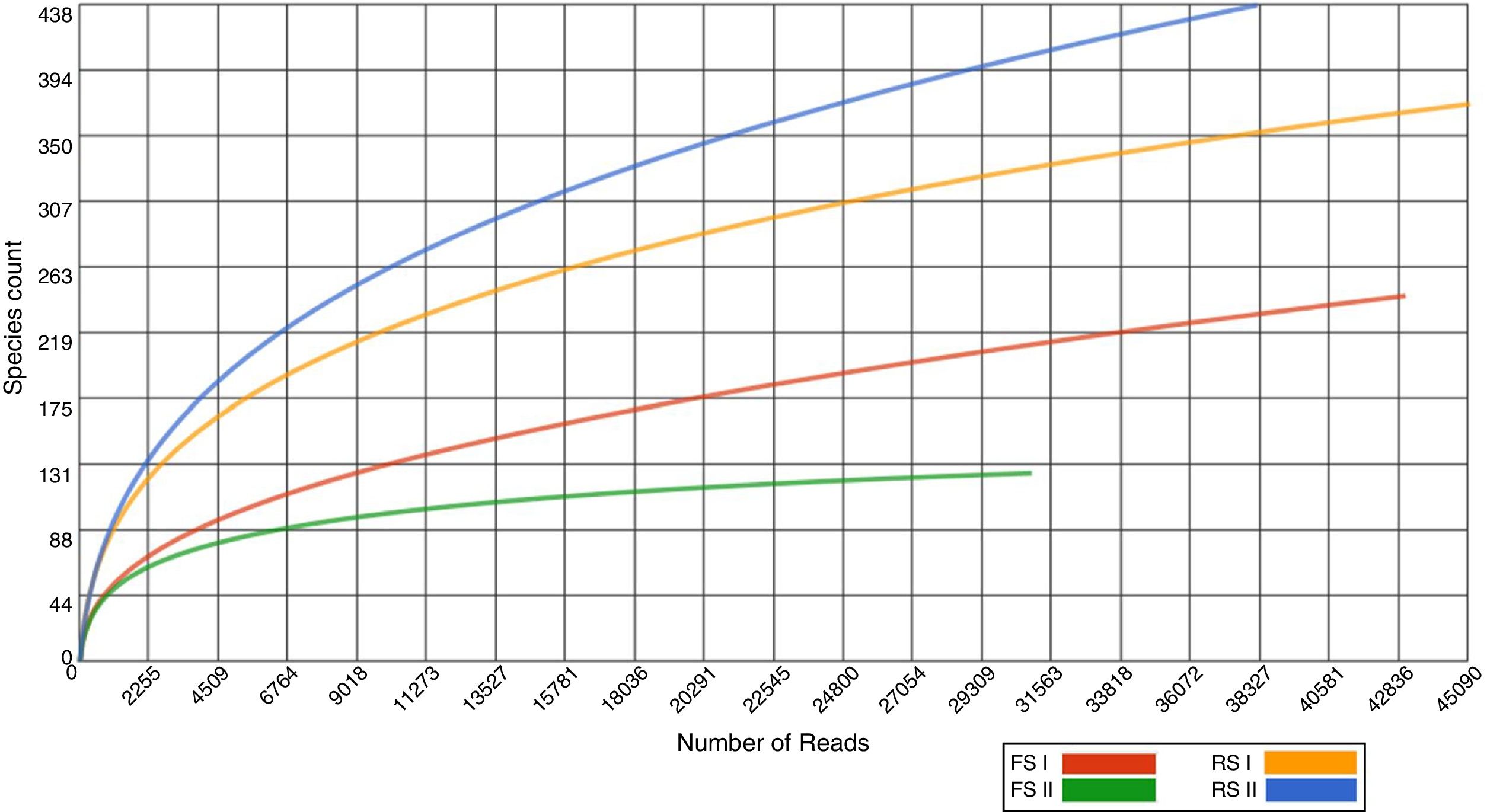
In total, 157,156 reads were generated with an average length of 214bp, and 69% of the sequences were annotated with the MG-Rast server (E value <10−5) after filtration (MG-Rast ID: mgm4560416.3, mgm4560417.3, mgm4560419.3, mgm4560420.3). We assessed the fraction of species sequenced from shale by-products by the rarefaction curves, which tended to approach the saturation plateau for FS samples, while RS samples showed that this by-product has not been exhaustively sampled (Fig. 1).

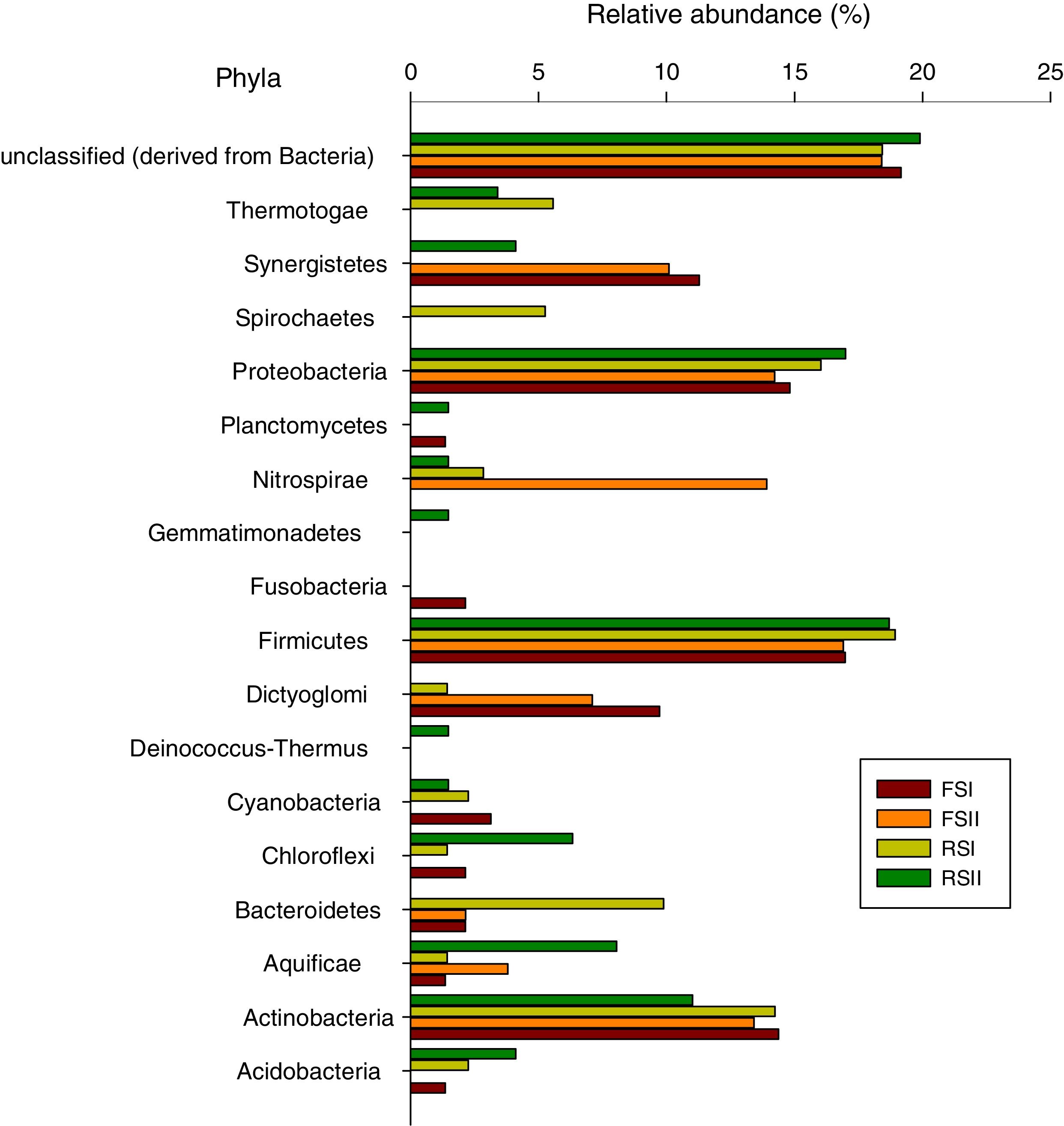
The results showed that 90% of the sequences were assigned to the domain bacteria. Of the 17 phyla detected in the shale by-products Firmicutes, Proteobacteria and Actinobacteria were the most abundant (Fig. 2). The FS had the lowest alpha diversity index with FSI at 6.26 and FSII at 6.86, while the α-diversity indices from RS (I and II) phylum were 24.34 and 15.87, respectively, showing that high bacterial diversity in the RS samples. The differences found between the two by-products could be partially explained by chemical composition or due to the microbial colonization after the pyrolysis process, since it involves high temperature treatment seems like this could be a sterilizing step with regards to the extant microorganisms. In areas surrounding the oil shale industry in the north-eastern part of Estonia were found that, even after 10 years, a few dominant populations and low diversity characterized the microbial community in this area.11,12
 of bacteria phyla found in shale by-product samples from Irati Formation, Brazil. Phylotypes defined at 95% similarity of sequence and analyzed in MG-Rast.")
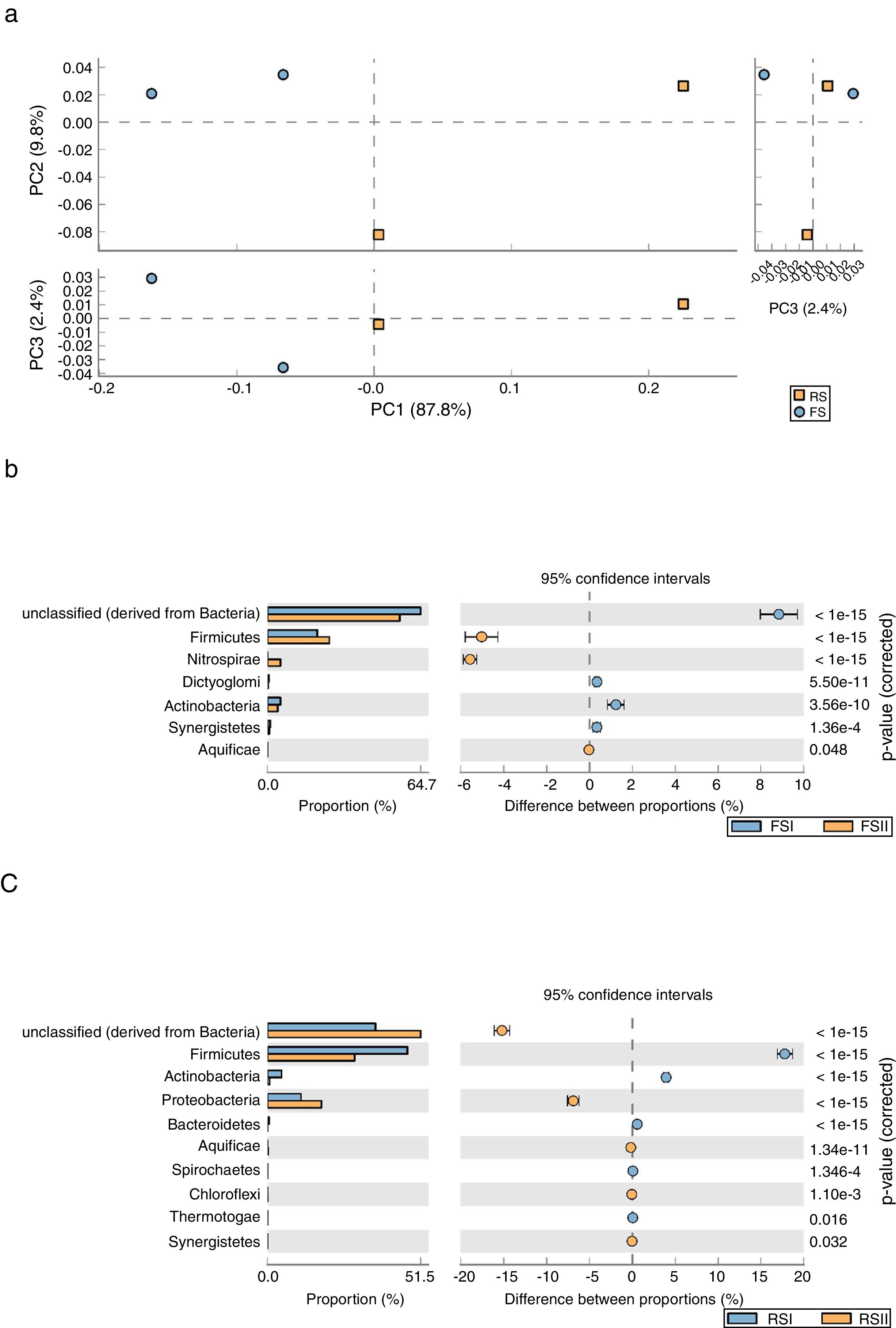
We employed STAMP software to examine the taxonomic profiles of enriched samples from the two RS and FS communities sampled from the mining area using a PCA plot and clustering analysis (heatmap) to compare the relative differences and/or similarities between by-products. The results showed that FS and RS by-products enriched communities are significantly different, which is represented by the upper and lower PCAs. The PCA plot indicated a robust distinction between the two communities from shale by-products. We observed a clear separation of groups in PCA1 and PCA2, where the two first principal components account for approximately 97.6% of the variation in the phylum group distribution. PCA take out a widely predominant component (PC1), accounting for 87.8% and of the variability in the data and clearly related to overall bacterial phyla communities from RS while PC2 that explained only 9.8% comprise FS community (Fig. 3A). A STAMP comparison at the phylum level reveals that Firmicutes and Nitrospirae were significantly overrepresented (P<1e−15) in the set of reads from FSII, while Actinobacteria and unclassified bacteria were overrepresented in FSI (Fig. 3B). The comparisons for RS show that Firmicutes and Actinobacteria were significantly overrepresented (P<1e−15) in the set of reads from RSI, while Proteobacteria and unclassified bacteria are overrepresented in the RSII (Fig. 3C).
 Principal components analysis (PCA) of phyla abundance from bacterial communities in Fine Shale (FS) and Retorted Shale (RS) by-products. STAMP comparisons of metagenomes: (B) the blue dot represents Fine Shale (FSI collected in 2008), and the orange represent FSII samples collected in 2011. (C) The orange dot represents Retorted Shale (RSII/2011), and the blue represent RSI/2008 samples.")
(A) Principal components analysis (PCA) of phyla abundance from bacterial communities in Fine Shale (FS) and Retorted Shale (RS) by-products. STAMP comparisons of metagenomes: (B) the blue dot represents Fine Shale (FSI collected in 2008), and the orange represent FSII samples collected in 2011. (C) The orange dot represents Retorted Shale (RSII/2011), and the blue represent RSI/2008 samples.
As indicated by the PCA plot, there were significant differences in the bacterial diversity found in the shale by-products, which can be due to differences in chemical characteristics resulting from the pyrolysis process suffered by RS to extract oil from rock. We consider those microorganisms present in RS samples are re-colonizers because, after oil extraction and cooling, this by-product is high in silicon content and also containing organic matter, micronutrients, hydrocarbons and other elements such as phosphorus, calcium, magnesium and sulfur. According to our previous studies with cultivable microorganisms there are higher diversity of fungi4 and bacteria in RS than in FS samples (data not published).
This study presents the first 16S rRNA gene massive sequence analysis of shale by-products from Brazil. PCR amplification of 16S rRNA from organisms which are not detected in plating method has provided an invaluable insight into our understanding of the community structure, particularly of communities inhabiting this environments, whose growth conditions may be difficult to mimic in the laboratory.13 The FSII show the smallest number of identified phyla, including Actinobacteria, Aquificae, Bacteroidetes, Dictyoglomi, Firmicutes, Nitrospirae, Proteobacteria and Synergistetes. FSI contained the phyla Acidobacteria, Actinobacteria, Aquificae, Bacteroidetes, Chloroflexi, Cyanobacteria, Dictyoglomi, Firmicutes, Fusobacteria, Planctomyces, Proteobacteria and Synergistetes. From the FSI and II samples, 19 and 18% of the total sequences remained unclassified (derived from bacteria), respectively. Thirteen phyla were identified from RSI, Acidobacteria, Actinobacteria, Aquificae, Bacteroidetes, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Dictyoglomi, Firmicutes, Nitrospirae, Proteobacteria, Spirochaetes and Thermotogae, and from RSII, Acidobacteria, Actinobacteria, Aquificae, Chloroflexi, Cyanobacteria, Deinococcus-Thermus, Firmicutes, Gemmatimonadetes, Nitrospirae, Planctomyces, Proteobacteria, Synergistetes and Thermotogae. On average, 18% and 20% of the total sequences from RSI and II were unclassified (derived from bacteria), respectively. When we compared our data obtained from Brazilian shale by-products with those from Li et al.14 from China, it were found 15 phylum common to both resources, among these Proteobacteria, Actinobacteria and Firmcutes were more dominant in both countries.
There is a general agreement that Firmicutes, Proteobacteria and Actinobacteria phylum, dominate all by-products, although some differences were observed in the number of reads, and they occurred in a greater number. Firmicutes was the most abundant phylum in all the samples and accounted for 30.42% of the total bacterial sequences. Proteobacteria and Actinobacteria were the second and third most abundant, corresponding to 10.87 and 3.94% of the reads, respectively. The predominance of Firmicutes, Proteobacteria and Actinobacteria was not surprising because these phyla have been found in other regions, including organic-rich black shales7,14 and acid mines.15 The dominance of the these three phyla was suggested as due to their adaptability to living in the metal-rich and extremely acidic shale environment when compared with other 30 different phyla found in a group of nine black shale samples in a depositional unit at Chengkou County, Southwest China.14 In Australia, high metal contents in soils overlying mineral deposits were discriminatory for a variety of bacterial taxa, including Acidobacteria, Bacilli, Betaproteobacteria, and Epsilonproteobacteria.16 However, this is also true for many other environments, like oil reservoirs or contaminated groundwater what seems that genera/species within these phyla are established in many diverse environments. Also Hill et al.17 based on an analysis for a broad range of environmental samples, suggested that actinobacterial community structure seems to be selected by human and animal activities.
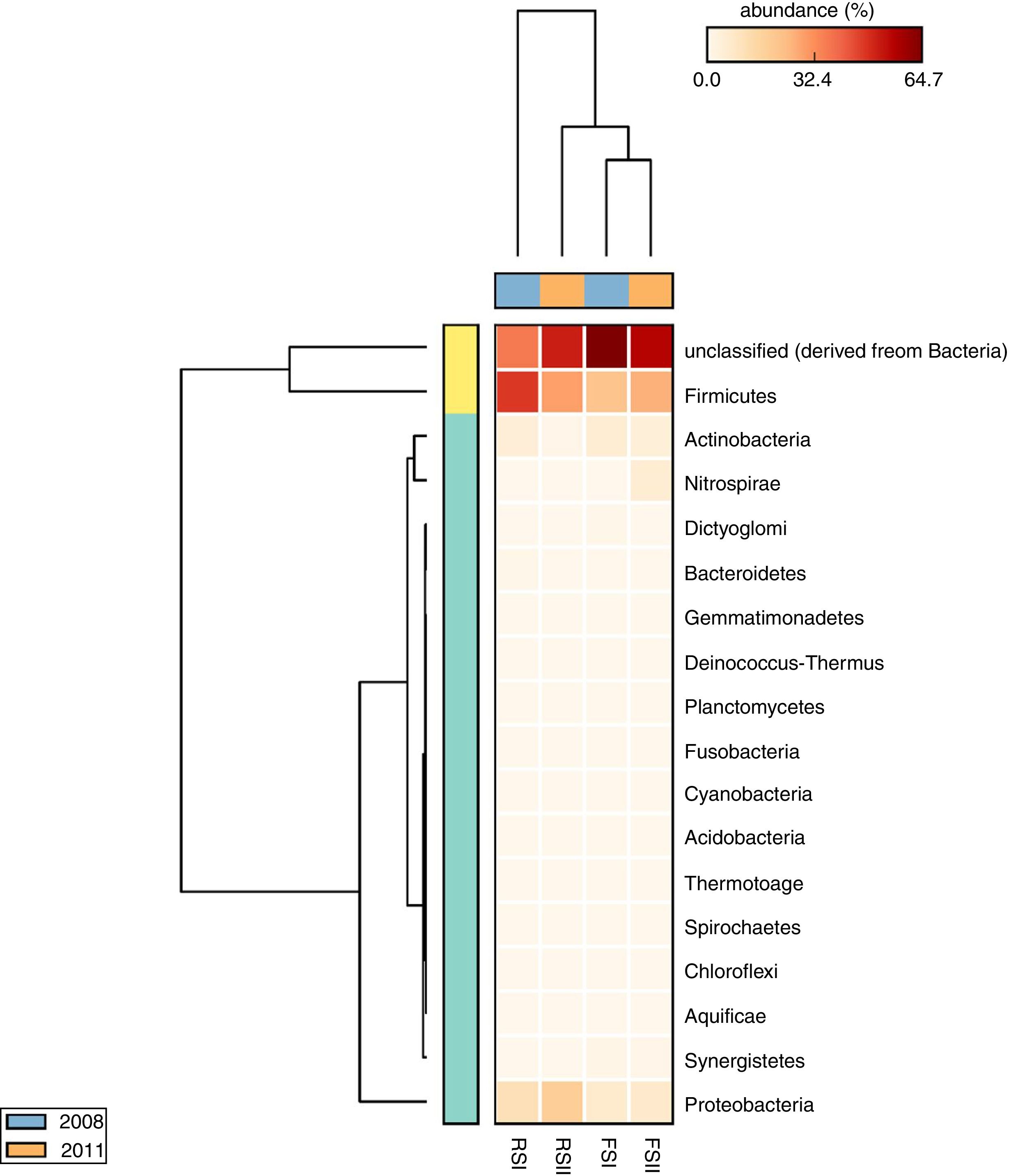
The taxonomic profiles of microbial communities when compared by cluster analysis based on a heatmap showed a clear separation between the phylum groups, confirming the differences in the by-products microbial communities (Fig. 4). The heatmap has two dendrograms, one indicating the similarity/dissimilarity among metagenomic samples, x-axis dendrogram, the horizontal orange (sampling in 2011) and blue (sampling in 2008) bars showing that when compared sampling there were differences only for RS and another indicating the similarity/dissimilarity among phyla, the y-axis dendrogram, where Firmicutes and unclassified bacteria group are separated from others phyla. We also observed that the FS bacterial communities are similar in their sampling times, while samples of RSI differ in their bacteria community composition.
 based on taxonomic profiles of microbial communities of shale by-products from the multiple comparison by Tukey-Krammer (p> or <0.05).")
In previous studies, as well as in our study, bacteria have been reported as indigenous to shale by-products.7,18,19 At times, relatively limited diversity has been observed, such as in the work performed by using denaturing gradient gel electrophoresis analysis to study bacteria distributed across the shale-sandstone at Cerro Negro, New Mexico, where the community was comprised of γ and δ-Proteobacteria, Flexibacter-Cytophaga-Bacteroides, sulfate-reducing bacteria and members of the Geobacteraceae family.20 For instance, indigenous bacteria play a prominent role in the weathering of black shale and in the biogeochemical cycles of elements in this rock.18,19
Our study provides the first 16S rRNA gene sequence analysis data about the microbial community present in oil shale by-products from Irati Formation, which will contribute to a better understanding of bacterial community present in this environment. Among the bacteria, Actinobacteria, Firmicutes and Proteobacteria dominate FS and RS at the phylum level. Deinococcus-Thermus, Gemmatimonadetes, Spirochaetes and Thermotogae were found only in RS samples. These results provide tools for studies and/or search of specific microorganisms to future applications.
Authors’ contributionsKCGPG, GML and DSA initiated and designed the study. DSA contributed with reagents/materials. KCGPG and GML performed the experiments. KCGPG, GML and DSA analyzed the data and wrote the paper. All authors read and approved the final manuscript.
Consent for publicationAll authors gave the consented for publication.
Ethics approval and consent of participationThe study has not involved any human or animal participation or data.
Conflicts of interestNo conflicts of interest declared.
The authors acknowledge financial support given by Petrobras SIX, São Mateus do Sul, PR – Agricultural Shale Project at Instituto Agronômico do Paraná (IAPAR). K.C.G.P. Goes acknowledges scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) at the Universidade Estadual de Londrina, Paraná, Brazil. D. S. Andrade acknowledges Fundação Araucária (Grant 42306/2014).