


In the previous study, we used genome shuffling to improve fengycin production of the original strain Bacillus amyloliquefaciens ES-2–4. After two rounds of genome shuffling, a high-yield recombinant FMB72 strain that exhibited 8.30-fold increase in fengycin production was obtained. In this study, comparative proteomic analysis of the parental ES-2–4 and genome-shuffled FMB72 strains was conducted to examine the differentially expressed proteins. In the shuffled strain FMB72, 50 differently expressed spots (p<0.05) were selected to be excised and analyzed using Matrix-Assisted Laser Desorption/Ionization Time of Flight/Time of Flight Mass Spectrometry, and finally 44 protein spots were confidently identified according to NCBI database. According to clusters of orthologous groups (COG) functional category analysis and related references, the differentially expressed proteins could be classified into several functional categories, including proteins involved in metabolism, energy generation and conversion, DNA replication, transcription, translation, ribosomal structure and biogenesis, cell motility and secretion, signal transduction mechanisms, general function prediction. Of the 44 identified proteins, signaling proteins ComA and Spo0A may positively regulate fengycin synthesis at transcriptional level. Taken together, the present study will be informative for exploring the exact roles of ComA and Spo0A in fengycin synthesis and explaining the molecular mechanism of fengycin synthesis.
Bacillus strains can produce many kinds of bioactive peptides synthesized non-ribosomally by a large multifunctional enzyme complex. Of these, fengycin specifically acting against filamentous fungi1 is biosynthesized by fengycin synthetase encompassing the five non-ribosomal peptide synthetases (NRPSs) Fen1-Fen5, respectively is coded by the gene fen A-E.2 Fengycin consists of a β-hydroxy fatty acid connected to the N-terminus of a decapeptide including four d-amino acid residues and the rare amino acid l-ornithine. The C-terminal residue of the peptide moiety is linked to the tyrosine residue at position 3, forming the branching point of the acylpeptide and the eight-membered cyclic lactone.3 Fengycin has potential applications in plant disease biocontrol,4 biomedicine, food5 and cosmetics6 industries. Therefore, it is particularly significant to improve fengycin production by industrial Bacillus strains.
Genome shuffling is an efficient approach for the rapid improvement of microbial phenotypes.7 We previously described the generation of a high-yield recombinant Bacillus amyloliquefaciens FMB72 strain that exhibited 8.30-fold increases in fengycin production, following two rounds of genome shuffling. Comparative research of synthetase gene expression was conducted between the parent strain and mutant strain using FQ (fluorescent quantitation) RT-PCR. Delta CT (threshold cycle) relative quantitation analysis indicated that fengycin synthetase gene (fenA) expression in the FMB72 strain was 12.77-fold greater than in the parent strain ES-2–4 at the transcriptional level.
However, the results only indirectly identified differences in fengycin synthetase gene at the transcriptional level. Because proteins execute molecular functions and are in charge of almost all the biochemical activities of the cell, a deep-dyed comprehension of biological systems requires the direct research of proteins. The proteomics technology based on two-dimensional electrophoresis, identification by MALDI-TOF/MS, and bioinformatics provides a good approach for large-scale proteomic analyses. In this research, the molecular mechanism of high-yield fengycin will be explored by comparative proteomics analysis of differentially expressed proteins between the parental and genome-shuffled strains.
Materials and methodsStrains and culture conditionsB. amyloliquefaciens ES-2–4 was the initial strain.8,9B. amyloliquefaciens FMB72 was the genome-shuffled mutant strain of B. amyloliquefaciens ES-2–4.10 The yield of fengycin increased by 8.30-fold compared to ES-2–4. These strains are preserved by the Key Laboratory of Food Processing and Quality Control of the Food Science and Technology College at Nanjing Agricultural University, Nanjing, China. B. amyloliquefaciens ES-2–4 was cultured in PDA (potato dextrose agar) media at 37°C. All microorganisms were conserved in BPY supplemented with 20% (v/v) glycerol and stored at −70°C. Seed medium (BPY) (beef extract 5.0g/L, peptone 10.0g/L, yeast extract paste 5.0g/L, NaCl 5.0g/L, glucose 10.0g/L) and fermentation medium (modified Landy) (l-sodium glutamate 4.0g/L, glucose 42.0g/L, KCl 0.5g/L, MgSO4 0.5g/L, CuSO4 0.16mg/L, KH2PO4 1.0g/L, MnSO4 5.0mg/L, FeSO4 0.15mg/L) were adjusted to pH 7.0.
Protein sample preparationThe strains were cultured at 30°C, 180rpm for 36h. Cells were harvested by centrifugation at 6000rpm for 5min at 4°C, and then washed 3 times with 20mmol/L Tris–HCl (pH 6.8). Cells were subsequently resuspended in lysis buffer containing 2mol/L thiourea, 7mol/L urea, 40mmol/L DTT, 4% (w/v) CHAPS, and 2% (v/v) pH 3–10 IPG buffer.11,12 Cells were cracked by sonication in an ultrasonic cell pulverizer (Ningbo Xin-zhi Biotechnology Co., China), equipped with a cup horn, for 45min on ice. Following ultrasonication, Nuclease Mix (GE Healthcare, Little Chalfont, United Kingdom) was added to a final concentration of 1% (v/v). The mixture was incubated for 1h at room temperature and then centrifuged for 30min at 13,000×g at 4°C. 2-D Quant kit (GE Healthcare) was used to assay the protein concentration, with bovine serum albumin as the standard.13 The samples were stored at −80°C until 2-DE.
2-DE analysis and stainingIn the first dimension, total whole-cell protein (250μg) was loaded onto the IPG strips (24cm, pH 4–7, GE Healthcare) which had been rehydrated 14h with 120mL rehydration solution (7mol/L urea, 2mol/L thiourea, 18mmol/L DTT, 2% Bio-Lyte, 2% (w/v) CHAPS, and 0.002% (w/v) bromophenol blue). Isoelectric focusing was performed on an EttanIPGphor 3 IEF system (GE Healthcare) for a total of 80kVh at 20°C. The voltage was set at 50V for 10h, 250V for 3h, 500V for 3h, 1000V for 1h, and 8000V for 1h, followed by 8000V until final volt-hours were reached. Subsequently, the strips were equilibrated for 15min in 2% (w/v) DTT in equilibration buffer (6mol/L urea, 75mmol/L Tris–HCl (pH 8.8), 30% (v/v) glycerol and 2% (w/v) SDS) followed by 15min in 2.5% (w/v) IAA in equilibration buffer. The strips were then transferred to 12.5% (w/v) SDS-polyacrylamide gels. The second dimension electrophoresis was carried out in an Ettan DALTII system (GE Healthcare) with a constant power of 5W per gel for the first 30min, followed by 12W per gel for 6.5–7.5h until the bromophenol blue front reached the bottom of the gels. The gels were placed into fixative solutions (10% acetic acid,40% methanol) overnight and then stained with 0.25% (w/v) silver nitrate.14 The biological replicates were performed for each treatment at least three times.
Image acquisition and data analysisThe silver-stained 2-DE gels were imaged by an ImageScanner (GE Healthcare), and analyzed on the Image Master 2D Elite software Version 2.00 (GE Healthcare). Images were properly cropped and optimized, and then gel-to-gel matching of the standard protein maps was performed. The spot detection parameters were optimized by checking different protein spots in certain regions of the gel and then automatically detected, followed by visual inspection for removal or addition of undetected spots. Spot detection was refined by manual spot edition when needed. The percentage volumes were used to designate the significant differentially expressed spots (at least two-fold increase/decrease and statistically significant as calculated by one-way ANOVA, p<0.05). Triplicate gels were used for each sample and the SD was calculated. Finally, only those protein spots that showed reproducible and changed more than 2-fold were considered to be differentially expressed proteins.
Protein in-gel digestionSpots showing changes statistically significant (p<0.05) and above a 2-fold threshold were excised from the gels and washed with double-distilled water and then transferred to sterilized Eppendorf tubes. Then, the protein spots were washed at room temperature with 25mmol/L NH4HCO3, followed by dehydration with 50% (v/v) acetonitrile (ACN) in 25mmol/L NH4HCO3. The proteins were then reduced with 10mmol/L DTT in 25mmol/L NH4HCO3 at 56°C for 1h, and then alkylated in 55mmol/L iodoacetamide in 25mmol/L NH4HCO3 for 45min at room temperature in darkness. The liquid was discarded and gel pieces were washed three times in 25mmol/L NH4HCO3, dehydrated in CAN and dried in a vacuum centrifuge. Gel pieces were then rehydrated in 25mmol/L NH4HCO3 containing 40ng trypsin, and incubated at 4°C for 1h. Excess liquid was discarded and gel plugs were incubated at 37°C overnight, with tubes inverted to keep gel pieces wet for sufficient enzymatic cleavage. Then, 5% (v/v) trifluoroacetic acid (TFA) was added and samples were incubated at 37°C for 1h. Supernatants were collected and the proteins were extracted twice by incubating the gel pieces in 8μL of 2.5% TFA in 50% ACN at 37°C for 1h. The resulting peptides were gathered and stored a −20°C until analysis.
Protein identification by MALDI-TOF/TOF and database searchSamples were air-dried and analyzed by a Biflex IV MALDI-TOF-MS (Bruker, Billerica, MA, USA). The N2 laser was operated at an accelerating voltage of 19kV with a wavelength of 337nm (3ns pulse length).
Data analysis was performed with the National Center for Biotechnology Information (NCBI) nr database using the MASCOT search program (Matrix Science, Boston, MA, USA). The following parameters were allowed: taxonomy restrictions to other firmicutes, 120ppm mass tolerance in MS, one missed cleavage, oxidation (M) as a variable modification and carbamidomethyl (C) as a fixed modification. The confidence in the peptide mass fingerprinting (PMF) matches (p<0.05) was based on the MOWSE score and confirmed by the accurate overlapping of the matched peptides with the major peaks of the mass spectrum. Only the best matches with high confidence levels were chosen when the software gave more than one eligible result.
qRT-PCR verificationThe total RNA were isolated from B. amyloliquefaciens cultures using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) and then treated with RNase-free DNase. First-strand cDNA was conversed from total RNA using an RT-PCR kit (Fermentas, Vilnius, Lithuania15). Real-time PCR was performed as described in the paper.16 Band intensities were normalized to the 16S rDNA transcript band for 2−ΔΔCT relative quantification. The B. amyloliquefaciens nucleotide sequences for these genes were obtained from the NCBI GenBank database. Primer pairs were designed from these sequences with Primer Premier 5.0 software (Applied Biosystems), the 16S rDNA primers used were F(5-CCTACGGGAGGCAGCAG-3) and R (5-ATTACCGCGGCT GCTGG-3), the comA primers were F (5-TCAAAGTGAGCAGGATCGGTTAA-3) and R (5-CTTCTGTACGGGAGCCGACAT-3) and spo0A primers were F (5-TTGCGGCG ATGAAGTGAATG-3) and R (5-CGATGGAAAGCTGCGGTGTA-3).
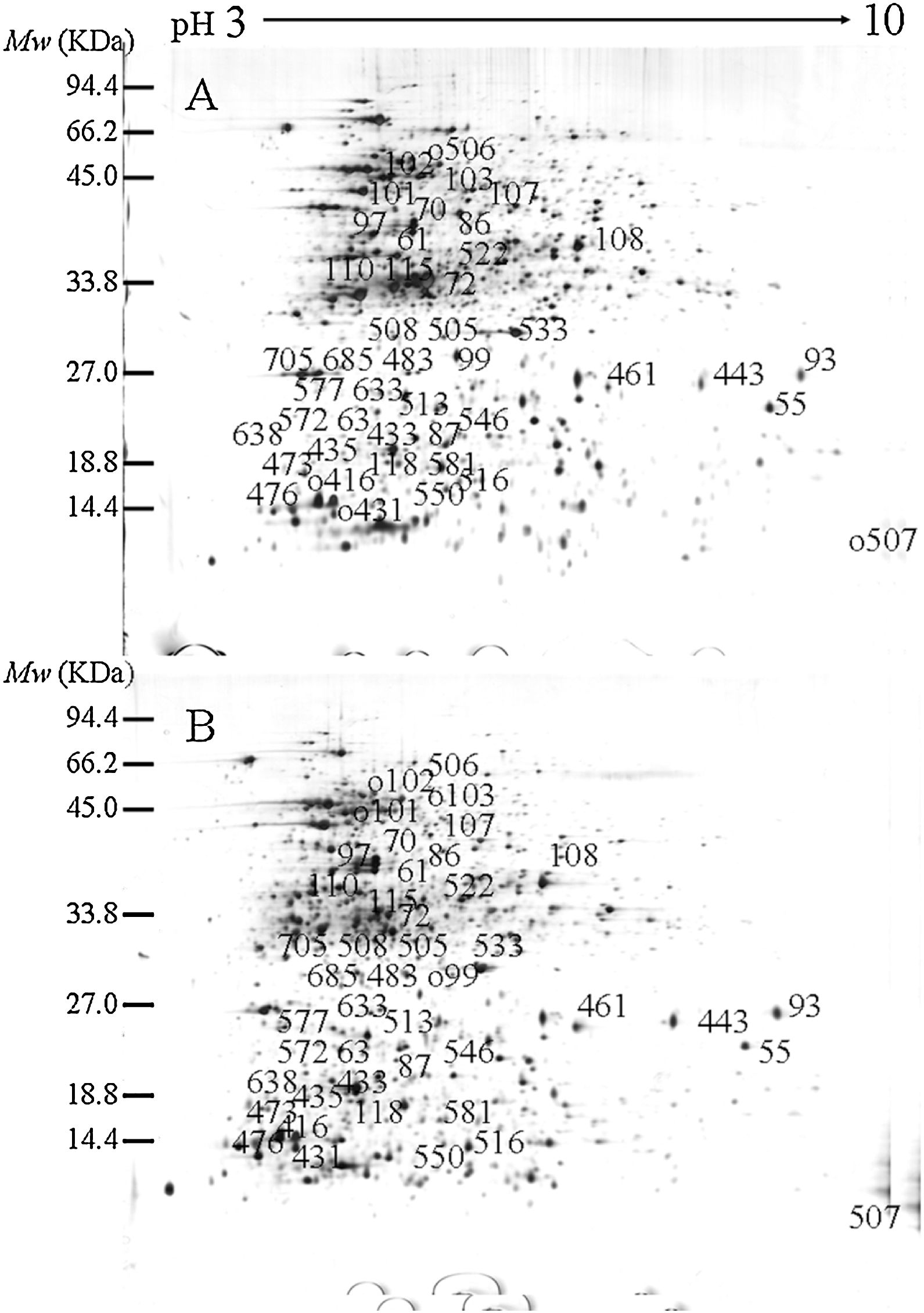
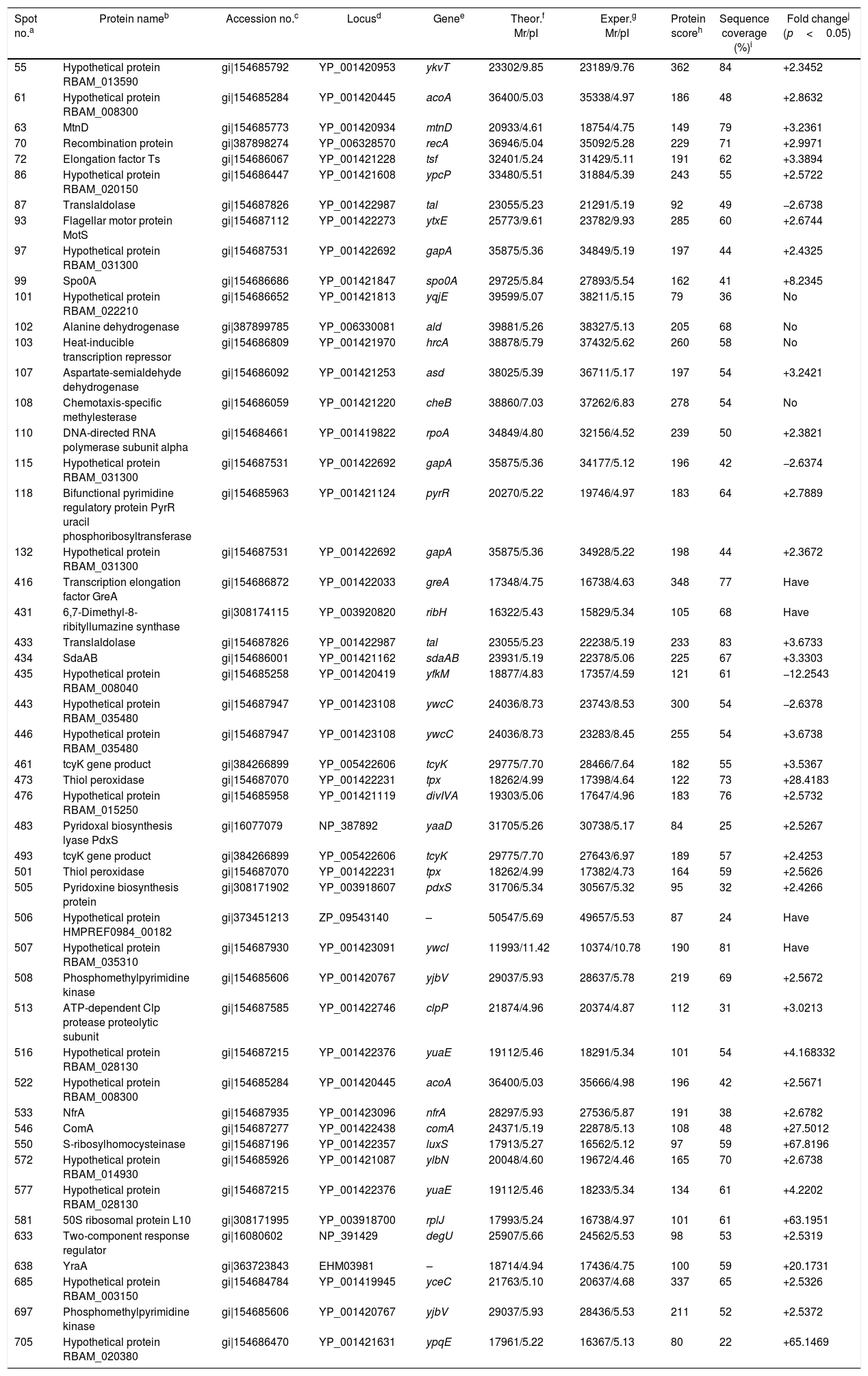
ResultsIdentification of differentially expressed proteinsTwo-DE profiles of soluble proteins were analyzed from parental (ES-2–4) and mutant (FMB72) strains. We found 50 protein spots that differed between the strains (Fig. 1). These 50 protein spots were identified by MALDI-TOF/MS analysis and their complete peptide fingerprints were gained. A search through the NCBI nr database using Mascot revealed that protein spots 87 and 433, 115 and 132, 443 and 446, 461 and 493, 473 and 501, 508 and 697 were the same proteins, meaning that a total of 44 proteins were successfully identified. In B. amyloliquefaciens FMB72, 37 proteins had increased expression, 4 proteins had decreased expression, 5 proteins appeared only in ES-2–4 and 4 proteins appeared only in FMB72 (Table 1).
 of B. amyloliquefaciens FMB72. (A) ES-2–4; (B) FMB72.")
Identification of differentially regulated cellular proteins (>2-fold change in expression) of B. amyloliquefaciens FMB72.
| Spot no.a | Protein nameb | Accession no.c | Locusd | Genee | Theor.f Mr/pI | Exper.g Mr/pI | Protein scoreh | Sequence coverage (%)i | Fold changej (p<0.05) |
|---|---|---|---|---|---|---|---|---|---|
| 55 | Hypothetical protein RBAM_013590 | gi|154685792 | YP_001420953 | ykvT | 23302/9.85 | 23189/9.76 | 362 | 84 | +2.3452 |
| 61 | Hypothetical protein RBAM_008300 | gi|154685284 | YP_001420445 | acoA | 36400/5.03 | 35338/4.97 | 186 | 48 | +2.8632 |
| 63 | MtnD | gi|154685773 | YP_001420934 | mtnD | 20933/4.61 | 18754/4.75 | 149 | 79 | +3.2361 |
| 70 | Recombination protein | gi|387898274 | YP_006328570 | recA | 36946/5.04 | 35092/5.28 | 229 | 71 | +2.9971 |
| 72 | Elongation factor Ts | gi|154686067 | YP_001421228 | tsf | 32401/5.24 | 31429/5.11 | 191 | 62 | +3.3894 |
| 86 | Hypothetical protein RBAM_020150 | gi|154686447 | YP_001421608 | ypcP | 33480/5.51 | 31884/5.39 | 243 | 55 | +2.5722 |
| 87 | Translaldolase | gi|154687826 | YP_001422987 | tal | 23055/5.23 | 21291/5.19 | 92 | 49 | −2.6738 |
| 93 | Flagellar motor protein MotS | gi|154687112 | YP_001422273 | ytxE | 25773/9.61 | 23782/9.93 | 285 | 60 | +2.6744 |
| 97 | Hypothetical protein RBAM_031300 | gi|154687531 | YP_001422692 | gapA | 35875/5.36 | 34849/5.19 | 197 | 44 | +2.4325 |
| 99 | Spo0A | gi|154686686 | YP_001421847 | spo0A | 29725/5.84 | 27893/5.54 | 162 | 41 | +8.2345 |
| 101 | Hypothetical protein RBAM_022210 | gi|154686652 | YP_001421813 | yqjE | 39599/5.07 | 38211/5.15 | 79 | 36 | No |
| 102 | Alanine dehydrogenase | gi|387899785 | YP_006330081 | ald | 39881/5.26 | 38327/5.13 | 205 | 68 | No |
| 103 | Heat-inducible transcription repressor | gi|154686809 | YP_001421970 | hrcA | 38878/5.79 | 37432/5.62 | 260 | 58 | No |
| 107 | Aspartate-semialdehyde dehydrogenase | gi|154686092 | YP_001421253 | asd | 38025/5.39 | 36711/5.17 | 197 | 54 | +3.2421 |
| 108 | Chemotaxis-specific methylesterase | gi|154686059 | YP_001421220 | cheB | 38860/7.03 | 37262/6.83 | 278 | 54 | No |
| 110 | DNA-directed RNA polymerase subunit alpha | gi|154684661 | YP_001419822 | rpoA | 34849/4.80 | 32156/4.52 | 239 | 50 | +2.3821 |
| 115 | Hypothetical protein RBAM_031300 | gi|154687531 | YP_001422692 | gapA | 35875/5.36 | 34177/5.12 | 196 | 42 | −2.6374 |
| 118 | Bifunctional pyrimidine regulatory protein PyrR uracil phosphoribosyltransferase | gi|154685963 | YP_001421124 | pyrR | 20270/5.22 | 19746/4.97 | 183 | 64 | +2.7889 |
| 132 | Hypothetical protein RBAM_031300 | gi|154687531 | YP_001422692 | gapA | 35875/5.36 | 34928/5.22 | 198 | 44 | +2.3672 |
| 416 | Transcription elongation factor GreA | gi|154686872 | YP_001422033 | greA | 17348/4.75 | 16738/4.63 | 348 | 77 | Have |
| 431 | 6,7-Dimethyl-8-ribityllumazine synthase | gi|308174115 | YP_003920820 | ribH | 16322/5.43 | 15829/5.34 | 105 | 68 | Have |
| 433 | Translaldolase | gi|154687826 | YP_001422987 | tal | 23055/5.23 | 22238/5.19 | 233 | 83 | +3.6733 |
| 434 | SdaAB | gi|154686001 | YP_001421162 | sdaAB | 23931/5.19 | 22378/5.06 | 225 | 67 | +3.3303 |
| 435 | Hypothetical protein RBAM_008040 | gi|154685258 | YP_001420419 | yfkM | 18877/4.83 | 17357/4.59 | 121 | 61 | −12.2543 |
| 443 | Hypothetical protein RBAM_035480 | gi|154687947 | YP_001423108 | ywcC | 24036/8.73 | 23743/8.53 | 300 | 54 | −2.6378 |
| 446 | Hypothetical protein RBAM_035480 | gi|154687947 | YP_001423108 | ywcC | 24036/8.73 | 23283/8.45 | 255 | 54 | +3.6738 |
| 461 | tcyK gene product | gi|384266899 | YP_005422606 | tcyK | 29775/7.70 | 28466/7.64 | 182 | 55 | +3.5367 |
| 473 | Thiol peroxidase | gi|154687070 | YP_001422231 | tpx | 18262/4.99 | 17398/4.64 | 122 | 73 | +28.4183 |
| 476 | Hypothetical protein RBAM_015250 | gi|154685958 | YP_001421119 | divIVA | 19303/5.06 | 17647/4.96 | 183 | 76 | +2.5732 |
| 483 | Pyridoxal biosynthesis lyase PdxS | gi|16077079 | NP_387892 | yaaD | 31705/5.26 | 30738/5.17 | 84 | 25 | +2.5267 |
| 493 | tcyK gene product | gi|384266899 | YP_005422606 | tcyK | 29775/7.70 | 27643/6.97 | 189 | 57 | +2.4253 |
| 501 | Thiol peroxidase | gi|154687070 | YP_001422231 | tpx | 18262/4.99 | 17382/4.73 | 164 | 59 | +2.5626 |
| 505 | Pyridoxine biosynthesis protein | gi|308171902 | YP_003918607 | pdxS | 31706/5.34 | 30567/5.32 | 95 | 32 | +2.4266 |
| 506 | Hypothetical protein HMPREF0984_00182 | gi|373451213 | ZP_09543140 | – | 50547/5.69 | 49657/5.53 | 87 | 24 | Have |
| 507 | Hypothetical protein RBAM_035310 | gi|154687930 | YP_001423091 | ywcI | 11993/11.42 | 10374/10.78 | 190 | 81 | Have |
| 508 | Phosphomethylpyrimidine kinase | gi|154685606 | YP_001420767 | yjbV | 29037/5.93 | 28637/5.78 | 219 | 69 | +2.5672 |
| 513 | ATP-dependent Clp protease proteolytic subunit | gi|154687585 | YP_001422746 | clpP | 21874/4.96 | 20374/4.87 | 112 | 31 | +3.0213 |
| 516 | Hypothetical protein RBAM_028130 | gi|154687215 | YP_001422376 | yuaE | 19112/5.46 | 18291/5.34 | 101 | 54 | +4.168332 |
| 522 | Hypothetical protein RBAM_008300 | gi|154685284 | YP_001420445 | acoA | 36400/5.03 | 35666/4.98 | 196 | 42 | +2.5671 |
| 533 | NfrA | gi|154687935 | YP_001423096 | nfrA | 28297/5.93 | 27536/5.87 | 191 | 38 | +2.6782 |
| 546 | ComA | gi|154687277 | YP_001422438 | comA | 24371/5.19 | 22878/5.13 | 108 | 48 | +27.5012 |
| 550 | S-ribosylhomocysteinase | gi|154687196 | YP_001422357 | luxS | 17913/5.27 | 16562/5.12 | 97 | 59 | +67.8196 |
| 572 | Hypothetical protein RBAM_014930 | gi|154685926 | YP_001421087 | ylbN | 20048/4.60 | 19672/4.46 | 165 | 70 | +2.6738 |
| 577 | Hypothetical protein RBAM_028130 | gi|154687215 | YP_001422376 | yuaE | 19112/5.46 | 18233/5.34 | 134 | 61 | +4.2202 |
| 581 | 50S ribosomal protein L10 | gi|308171995 | YP_003918700 | rplJ | 17993/5.24 | 16738/4.97 | 101 | 61 | +63.1951 |
| 633 | Two-component response regulator | gi|16080602 | NP_391429 | degU | 25907/5.66 | 24562/5.53 | 98 | 53 | +2.5319 |
| 638 | YraA | gi|363723843 | EHM03981 | – | 18714/4.94 | 17436/4.75 | 100 | 59 | +20.1731 |
| 685 | Hypothetical protein RBAM_003150 | gi|154684784 | YP_001419945 | yceC | 21763/5.10 | 20637/4.68 | 337 | 65 | +2.5326 |
| 697 | Phosphomethylpyrimidine kinase | gi|154685606 | YP_001420767 | yjbV | 29037/5.93 | 28436/5.53 | 211 | 52 | +2.5372 |
| 705 | Hypothetical protein RBAM_020380 | gi|154686470 | YP_001421631 | ypqE | 17961/5.22 | 16367/5.13 | 80 | 22 | +65.1469 |
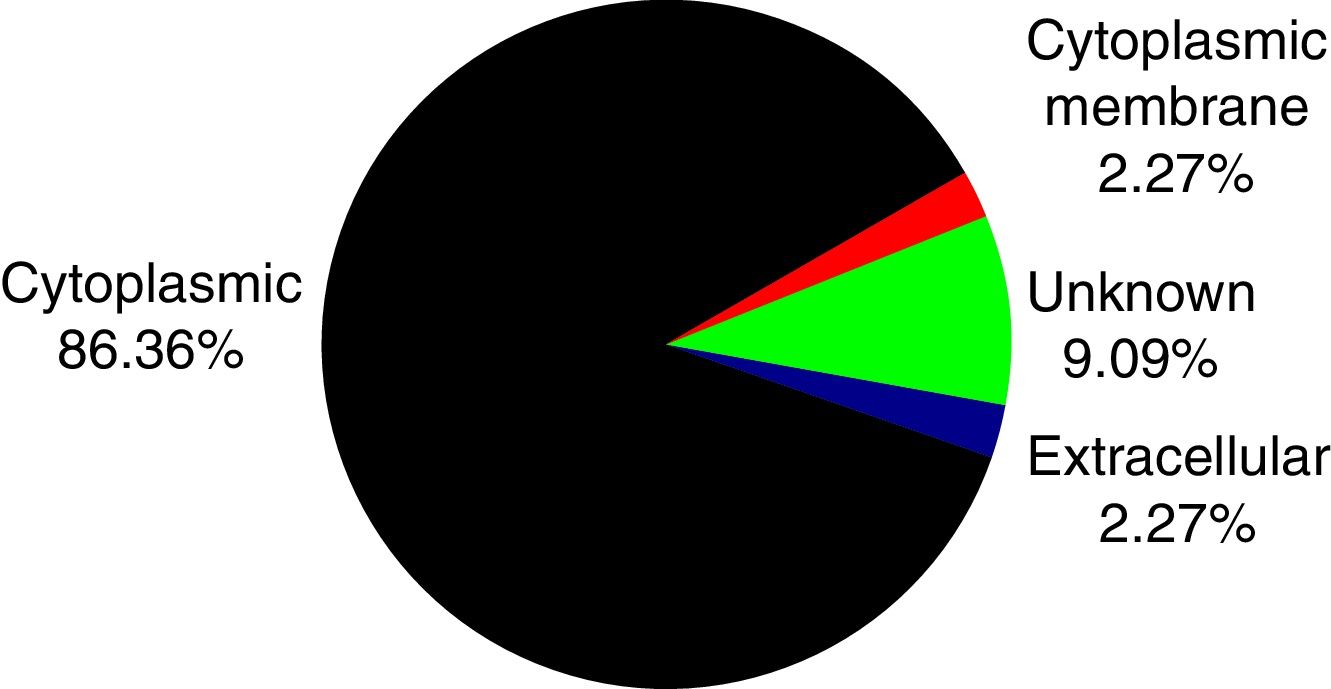
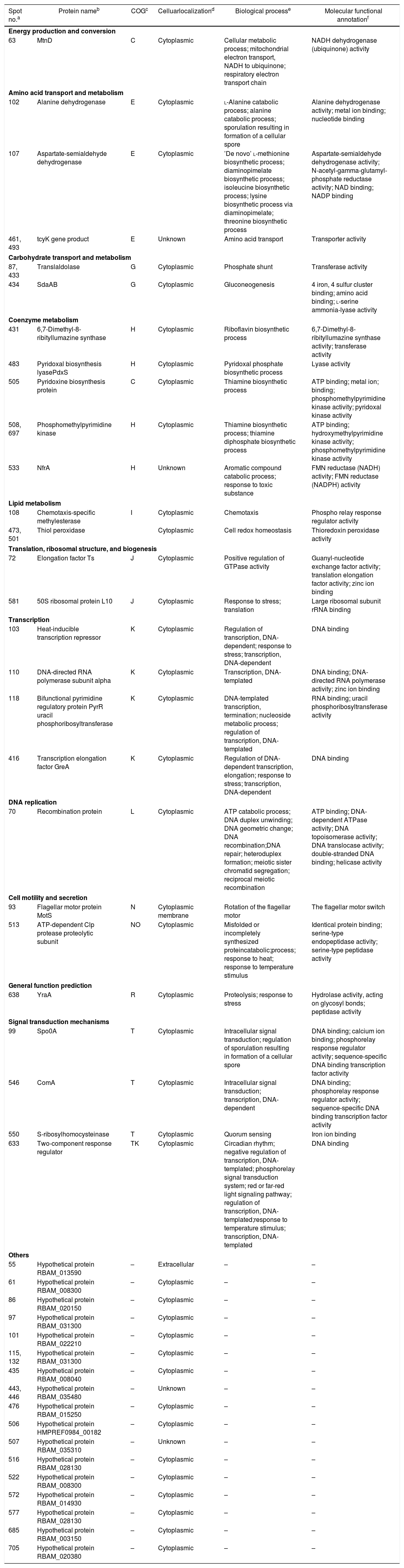
PSORTb tool version 3.0.2 (http://www.psort.org/psortb/index.html) was used to predict the cellular localization of the 44 identified proteins (Table 2). Thirty-nine proteins were found to be located at cytoplasm, one protein was in cytoplasmic membrane, one protein was extracellular, and four proteins had an unknown cellular location (Fig. 2).
Cellular localization and function of differentially regulated cellular proteins (>2-fold change in expression) of B. amyloliquefaciens FMB72.
| Spot no.a | Protein nameb | COGc | Celluarlocalizationd | Biological processe | Molecular functional annotationf |
|---|---|---|---|---|---|
| Energy production and conversion | |||||
| 63 | MtnD | C | Cytoplasmic | Cellular metabolic process; mitochondrial electron transport, NADH to ubiquinone; respiratory electron transport chain | NADH dehydrogenase (ubiquinone) activity |
| Amino acid transport and metabolism | |||||
| 102 | Alanine dehydrogenase | E | Cytoplasmic | l-Alanine catabolic process; alanine catabolic process; sporulation resulting in formation of a cellular spore | Alanine dehydrogenase activity; metal ion binding; nucleotide binding |
| 107 | Aspartate-semialdehyde dehydrogenase | E | Cytoplasmic | ’De novo’ l-methionine biosynthetic process; diaminopimelate biosynthetic process; isoleucine biosynthetic process; lysine biosynthetic process via diaminopimelate; threonine biosynthetic process | Aspartate-semialdehyde dehydrogenase activity; N-acetyl-gamma-glutamyl-phosphate reductase activity; NAD binding; NADP binding |
| 461, 493 | tcyK gene product | E | Unknown | Amino acid transport | Transporter activity |
| Carbohydrate transport and metabolism | |||||
| 87, 433 | Translaldolase | G | Cytoplasmic | Phosphate shunt | Transferase activity |
| 434 | SdaAB | G | Cytoplasmic | Gluconeogenesis | 4 iron, 4 sulfur cluster binding; amino acid binding; l-serine ammonia-lyase activity |
| Coenzyme metabolism | |||||
| 431 | 6,7-Dimethyl-8-ribityllumazine synthase | H | Cytoplasmic | Riboflavin biosynthetic process | 6,7-Dimethyl-8-ribityllumazine synthase activity; transferase activity |
| 483 | Pyridoxal biosynthesis lyasePdxS | H | Cytoplasmic | Pyridoxal phosphate biosynthetic process | Lyase activity |
| 505 | Pyridoxine biosynthesis protein | C | Cytoplasmic | Thiamine biosynthetic process | ATP binding; metal ion; binding; phosphomethylpyrimidine kinase activity; pyridoxal kinase activity |
| 508, 697 | Phosphomethylpyrimidine kinase | H | Cytoplasmic | Thiamine biosynthetic process; thiamine diphosphate biosynthetic process | ATP binding; hydroxymethylpyrimidine kinase activity; phosphomethylpyrimidine kinase activity |
| 533 | NfrA | H | Unknown | Aromatic compound catabolic process; response to toxic substance | FMN reductase (NADH) activity; FMN reductase (NADPH) activity |
| Lipid metabolism | |||||
| 108 | Chemotaxis-specific methylesterase | I | Cytoplasmic | Chemotaxis | Phospho relay response regulator activity |
| 473, 501 | Thiol peroxidase | Cytoplasmic | Cell redox homeostasis | Thioredoxin peroxidase activity | |
| Translation, ribosomal structure, and biogenesis | |||||
| 72 | Elongation factor Ts | J | Cytoplasmic | Positive regulation of GTPase activity | Guanyl-nucleotide exchange factor activity; translation elongation factor activity; zinc ion binding |
| 581 | 50S ribosomal protein L10 | J | Cytoplasmic | Response to stress; translation | Large ribosomal subunit rRNA binding |
| Transcription | |||||
| 103 | Heat-inducible transcription repressor | K | Cytoplasmic | Regulation of transcription, DNA-dependent; response to stress; transcription, DNA-dependent | DNA binding |
| 110 | DNA-directed RNA polymerase subunit alpha | K | Cytoplasmic | Transcription, DNA-templated | DNA binding; DNA-directed RNA polymerase activity; zinc ion binding |
| 118 | Bifunctional pyrimidine regulatory protein PyrR uracil phosphoribosyltransferase | K | Cytoplasmic | DNA-templated transcription, termination; nucleoside metabolic process; regulation of transcription, DNA-templated | RNA binding; uracil phosphoribosyltransferase activity |
| 416 | Transcription elongation factor GreA | K | Cytoplasmic | Regulation of DNA-dependent transcription, elongation; response to stress; transcription, DNA-dependent | DNA binding |
| DNA replication | |||||
| 70 | Recombination protein | L | Cytoplasmic | ATP catabolic process; DNA duplex unwinding; DNA geometric change; DNA recombination;DNA repair; heteroduplex formation; meiotic sister chromatid segregation; reciprocal meiotic recombination | ATP binding; DNA-dependent ATPase activity; DNA topoisomerase activity; DNA translocase activity; double-stranded DNA binding; helicase activity |
| Cell motility and secretion | |||||
| 93 | Flagellar motor protein MotS | N | Cytoplasmic membrane | Rotation of the flagellar motor | The flagellar motor switch |
| 513 | ATP-dependent Clp protease proteolytic subunit | NO | Cytoplasmic | Misfolded or incompletely synthesized proteincatabolic;process; response to heat; response to temperature stimulus | Identical protein binding; serine-type endopeptidase activity; serine-type peptidase activity |
| General function prediction | |||||
| 638 | YraA | R | Cytoplasmic | Proteolysis; response to stress | Hydrolase activity, acting on glycosyl bonds; peptidase activity |
| Signal transduction mechanisms | |||||
| 99 | Spo0A | T | Cytoplasmic | Intracellular signal transduction; regulation of sporulation resulting in formation of a cellular spore | DNA binding; calcium ion binding; phosphorelay response regulator activity; sequence-specific DNA binding transcription factor activity |
| 546 | ComA | T | Cytoplasmic | Intracellular signal transduction; transcription, DNA-dependent | DNA binding; phosphorelay response regulator activity; sequence-specific DNA binding transcription factor activity |
| 550 | S-ribosylhomocysteinase | T | Cytoplasmic | Quorum sensing | Iron ion binding |
| 633 | Two-component response regulator | TK | Cytoplasmic | Circadian rhythm; negative regulation of transcription, DNA-templated; phosphorelay signal transduction system; red or far-red light signaling pathway; regulation of transcription, DNA-templated;response to temperature stimulus; transcription, DNA-templated | DNA binding |
| Others | |||||
| 55 | Hypothetical protein RBAM_013590 | – | Extracellular | – | – |
| 61 | Hypothetical protein RBAM_008300 | – | Cytoplasmic | – | – |
| 86 | Hypothetical protein RBAM_020150 | – | Cytoplasmic | – | – |
| 97 | Hypothetical protein RBAM_031300 | – | Cytoplasmic | – | – |
| 101 | Hypothetical protein RBAM_022210 | – | Cytoplasmic | – | – |
| 115, 132 | Hypothetical protein RBAM_031300 | – | Cytoplasmic | – | – |
| 435 | Hypothetical protein RBAM_008040 | – | Cytoplasmic | – | – |
| 443, 446 | Hypothetical protein RBAM_035480 | – | Unknown | – | – |
| 476 | Hypothetical protein RBAM_015250 | – | Cytoplasmic | – | – |
| 506 | Hypothetical protein HMPREF0984_00182 | – | Cytoplasmic | – | – |
| 507 | Hypothetical protein RBAM_035310 | – | Unknown | – | – |
| 516 | Hypothetical protein RBAM_028130 | – | Cytoplasmic | – | – |
| 522 | Hypothetical protein RBAM_008300 | – | Cytoplasmic | – | – |
| 572 | Hypothetical protein RBAM_014930 | – | Cytoplasmic | – | – |
| 577 | Hypothetical protein RBAM_028130 | – | Cytoplasmic | – | – |
| 685 | Hypothetical protein RBAM_003150 | – | Cytoplasmic | – | – |
| 705 | Hypothetical protein RBAM_020380 | – | Cytoplasmic | – | – |
Protein name in the National Center for Biotechnology Information (NCBI) database for B. amyloliquefaciens.
Biological process was assigned according to the protein knowledge base (www.uniprot.org) for B. amyloliquefaciens.
Molecular functional annotation was assigned according to the protein knowledge base (www.uniprot.org) for B. amyloliquefaciens.

Experimentally identified proteins were functionally characterized by clusters of orthologous groups (COG) analysis (Table 2). The separated proteins were chiefly divided into the following categories: energy production and conversion (C), amino acid transport and metabolism (E), carbon transport and metabolism (G), coenzyme metabolism (H), lipid metabolism (I), translation, ribosomal structure, and biosynthesis (J), transcription (K), DNA replication (L), cell motility and secretion (N), general function prediction (R), signal transduction mechanisms (T) and not included in the COG classification (–). To determine the mechanism of increased antimicrobial peptide yield from B. amyloliquefaciens FMB72, biological process and molecular function data were acquired from the UniProKB (www.uniprot.org) database.
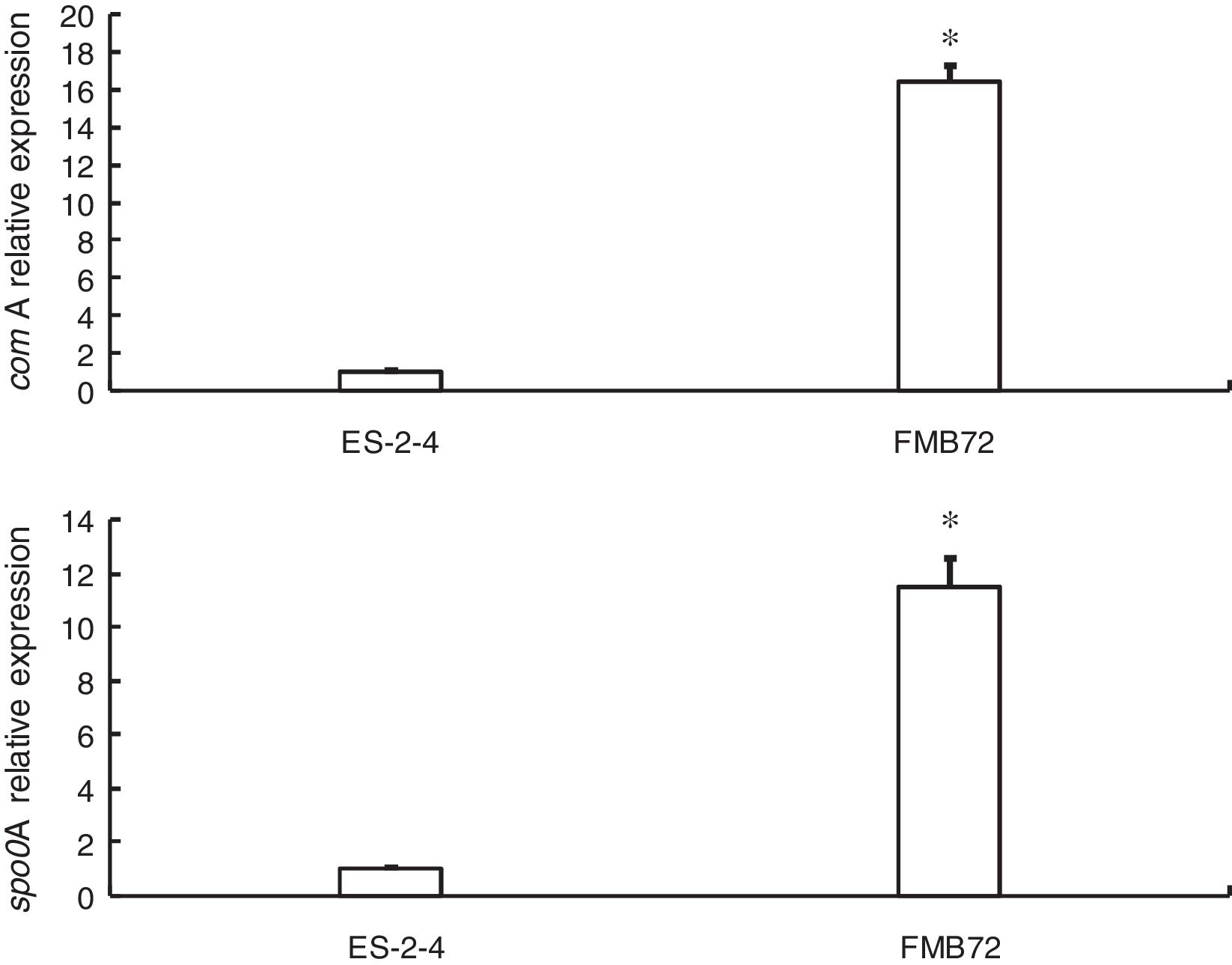
Gene expression verification by qRT-PCRExpression of the two genes encoding differentially expressed proteins related to fengycin synthesis (comA, spo0A) was analyzed by qRT-PCR analysis of mRNA from FMB72. The mRNA expression profiles of these genes are shown in Fig. 3. The mRNA levels of comA and spo0A were upregulated1 5.8 and 12.1 fold in FMB72. The upregulated expression of comA and spo0A mRNA in FMB72 agreed with their protein levels.
Discussion between the parental strain ES-2–4 and recombination strain FMB72.")
The majority of the identified proteins in this experiment are related to energy production and conversion, amino acid transport and metabolism, carbon transport and metabolism, coenzyme metabolism, lipid metabolism, translation, ribosomal structure, and biosynthesis, transcription, DNA replication, cell motility and secretion, general function prediction, signal transduction mechanisms, and not included in the COG classification. These proteins are analyzed as the following.
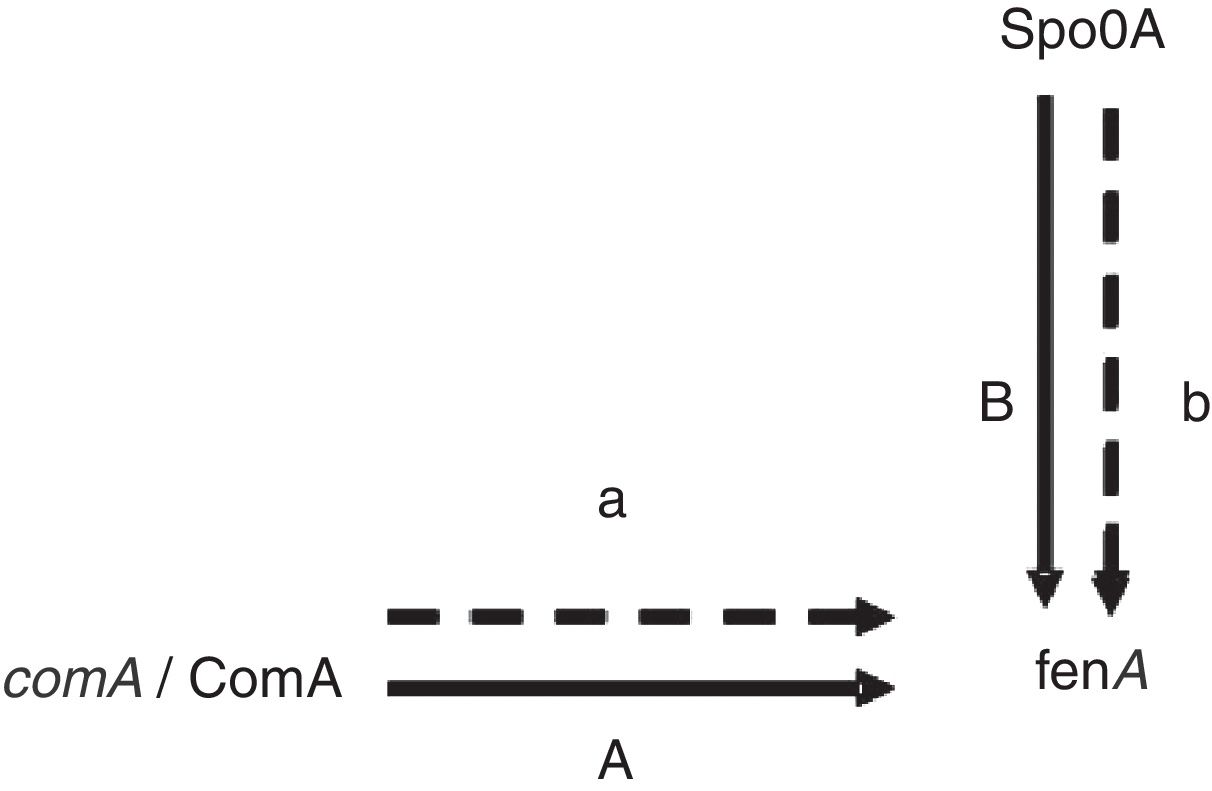
Proteins related to signal transduction mechanismsThere are four proteins involved in signal transduction mechanisms. The prokaryotes are mainly regulated at the transcriptional level. The activator protein bind the sequences close to promoter, the affinity enhancement of RNA polymerase with the promoter, and RNA polymerase activity augmentation. The repressor protein can hinder gene transcription by binding manipulation sequence. Spo0A is a very important regulatory protein in bacterial gene regulation system17 and the main control fact or of biofilm formation.18 It can control the opening and closing of downstream genes through phosphorylation and dephosphorylation. As previously mentioned Spo0A19 (upregulated) and ComA20 (upregulated) as a transcription factor, it is speculated that the expression change and fengycin production increase in the high-yield strain are closely related (Fig. 4). Antibiotics synthesis is closely related to the biofilm formation and the growth of the spore. It is showed that lipopeptide biosynthesis, biofilm formation and the growth of spores in the same metabolic network. Bacillus sporulation and biofilm formation are governed by the regulatory protein Spo0A. The upregulation of Spo0A is possible helpful to the formation of biofilm. It is reported that antibiotic is produced in a biofilm. Thereby, it is very well understood that the synthesis of fengycin is modulated by Spo0A. ComA could also work through modulating transition state gene expression. It is assumed that ComA acts as a transcriptional regulatory protein, and can directly bind to the fen promoter, thus promoting the synthesis and secretion of fengycin. The further functional verification of transcription proteins will be carried out in subsequent experiments.
 regulation. The solid lines show the regulation has been confirmed in the literature, the dash lines show the regulation speculated in this study.")
S-ribosylhomocysteinase upregulated in the recombination strain, has lyase activity, which is both combined with the iron ions and related to quorum sensing.21
Metabolism-related proteinsWe identified 12 differentially expressed proteins related to the metabolism of lipids, carbohydrates, coenzymes, and amino acids. The synthesis levels of key enzymes involved in glycolysis and the pentose phosphate pathway of glucose metabolism were higher in the genome-shuffled strain. The putative implications of these increases are described below. Transaldolase in the non-oxidative stage of the pentose phosphate pathway can catalyze the reaction between glyceraldehyde-3-phosphate and 7-sedoheptulose monophosphate to generate 4-phosphate erythrose and fructose 6-phosphate. NADP produced in the pentose phosphate pathway could provide reducing power for the biosynthesis reaction, while pentose phosphate generated in this pathway could then participate in nucleic acid metabolism. SdaAB can enhance gluconeogenesis, thus amino acids into sugar may be the main pathway of amino acid metabolism.
Alanine dehydrogenase plays an important role in amino acid transport and metabolism. Because of this function, it was widely believed that Aspartate-semialdehyde dehydrogenase played an important regulatory function in a series of pathological and physiological processes, has aspartate-semialdehyde dehydrogenase activity, and catalyzes the synthesis of aspartate. We speculate that the increased fengycin production by strain FMB72 is a result of increases in this enzyme.
Furthermore, expression levels of key enzymes in lipid metabolism and coenzyme metabolism processes were also raised. The upregulation of chemotaxis-specific methylesterase and thiol peroxidaseimprove lipid metabolism, and 6,7-dimethyl-8-ribityllumazine synthase, pyridoxal biosynthesis lyase PdxS, phosphomethylpyrimidine kinase, NfrA, pyridoxine biosynthesis protein would greatly improve the metabolic activities of coenzyme in the recombination strain. Thus, we speculate that the increase in fengycin yield accompanies the increased synthesis of key enzymes of the glycolysis and pentose phosphate pathways. Additionally, the abundance of alanine dehydrogenase and other key enzymes in amino acid metabolism likely enhances fengycin production.
Proteins related to energy generation and conversionOne of the identified protein (MtnD) was related to energy production and conversion, which is upregulated in the recombination strain. MtnD is in respiratory electron transport chain with NADH dehydrogenase (ubiquinone) activity. The enhancement of synthesis of enzymes associated with energy production and conversion may cause the increase of the fengycin yield indirectly.
Proteins related to DNA replicationThe DNA replication-related protein, recombination protein is upregulated with the function of DNA duplex unwinding, DNA recombination, DNA repair and heteroduplex formation. Thus, the capability of DNA replication is improved in fengycin high-yield strain FMB72.
Proteins related to transcriptionThe three proteins related to transcription, DNA-directed RNA polymerase subunit alpha, bifunctional pyrimidine regulatory protein PyrR uracil phosphoribosyltransferase and transcription elongation factor GreA are all up-regulated. The heat-inducible transcription repressor is down-regulated. Therefore, the level of transcription is strengthened in the shuffled strain.
Proteins related to translation, ribosomal structure, and biogenesisThere are two proteins related to translation, ribosomal structure, and biogenesis. One is elongation factor Ts, the other is 50S ribosomal protein L10.
Translation elongation factor Ts belong to the protein elongation factor family and is related to protein synthesis. The elongation factor Ts is one of the three elongation factors in prokaryotes and necessary for prokaryotic protein synthesis.
Ribosome is the place of protein biosynthesis. Ribosome size is demonstrated by the sedimentation coefficient S. There are approximately 20,000 ribosomes in a eugenic bacteria, wherein the proteins account for 10% of the total cellular proteins, rRNA account for 80% of the total cellular RNA. In prokaryote 70S ribosome, the 30 S subunit contain 22 kinds of the ribosomal protein, the 50 S subunit contain 34 kinds of the ribosomal protein, accounting for 35% of the ribosome. Ts and methionine aminopeptidase play an important role in protein processing. For this reason, the upregulation of two proteins in the recombination strain will more effectively ensure the process of protein translation.
Proteins related to cell motility and secretionThere are two proteins involved in cell motility and secretion, flagellar motor protein MotS and ATP-dependent Clp protease proteolytic subunit. In addition, the upregulation of the two proteins can improve the cell motility and secretion in FMB72.
Proteins related to general function prediction and hypothetical proteinsThere is one protein, YraA related to general function prediction. 17 spots subjected to mass spectrometry are identified as hypothetical proteins. They may play important roles directly and/or indirectly in response to fengycin synthesis. Thus, the more additional experiments are needed to gain the related protein function message in fengycin synthesis process.
ConclusionsAll these indicated that the metabolic capability of mutant was improved by the genome shuffling. We obtained two metabolic proteins in the database for which we are uncertain about their specific function. They are ComA (spot 546) and Spo0A (spot 99), respectively. They may play important roles directly and/or indirectly in response to fengycin synthesis.
Conflicts of interestThe authors declare no conflicts of interest.
This study was supported by the National Natural Science Foundation of China (No. 31571887).