








Equine influenza is one of the major respiratory infectious diseases in horses. An equine influenza virus outbreak was identified in vaccinated and unvaccinated horses in a veterinary school hospital in São Paulo, SP, Brazil, in September 2015. The twelve equine influenza viruses isolated belonged to Florida Clade 1. The hemagglutinin and neuraminidase amino acid sequences were compared with the recent isolates from North and South America and the World Organisation for Animal Health recommended Florida Clade 1 vaccine strain. The hemagglutinin amino acid sequences had nine substitutions, compared with the vaccine strain. Two of them were in antigenic site A (A138S and G142R), one in antigenic site E (R62K) and another not in antigenic site (K304E). The four substitutions changed the hydrophobicity of hemagglutinin. Three distinct genetic variants were identified during the outbreak. Eleven variants were found in four quasispecies, which suggests the equine influenza virus evolved during the outbreak. The use of an out of date vaccine strain or updated vaccines without the production of protective antibody titers might be the major contributing factors on virus dissemination during this outbreak.
Equine influenza (EI) leads to epithelial destruction in the upper respiratory tract of horses, resulting in fever, coughing, nasal discharge, performance impairment and occasionally pneumonia.1
The classification of influenza A viruses is based on two major surface antigens, hemagglutinin (HA) and neuraminidase (NA) and are related to virus infection and propagation.1 Minor changes in amino acid constitution can result in different antigenicity and lead to immune scape.2
Equine influenza viruses (EIVs) (H3N8) were initially assigned to a single cluster3 and evolved into two sublineages, American and Eurasian, according to the geographic region.4 The American lineage diverged into South American, Kentucky and Florida sublineages.5 The strains of the Florida sublineage had amino acid mutations in the hemagglutinin 1 (HA1) subunit and this lineage diverged into Florida Clade 1 (FC1), with the substitutions A78V and S159N, and Florida Clade 2 (FC2),6,7 represented by A/equine/South Africa/4/2003 and A/equine/Richmond/1/07, respectively.7 The lineages FC1 and FC2 have been identified in outbreaks worldwide and are the predominant circulating EIVs.8–16
EIV is considered endemic in Brazil, where the first outbreak caused by a H3N8 virus was reported in 1963,16–22 while the most recent H7N7 EIV outbreak happened in 1976.23,24 Outbreaks of H3N8 occurred in this country in 200825 and 2010,26 but data on the lineages to which the viruses belonged were not published.25,26 H3N8 EIVs isolated from a further outbreak in 2012 in the states of São Paulo and Rio Grande do Sul belonged to FC116,22 and, regardless of several substitutions in the HA1 subunit, no major differences in antigenicity were found in comparison with the Florida 1 vaccine strain A/equine/South Africa/4/2003.16
The aims of this study were (a) to describe an EIV outbreak in a veterinary school hospital in Brazil in 2015 that affected both vaccinated and unvaccinated horses; (b) to characterize the HA1 and neuraminidase (NA) amino acid sequences in comparison with the vaccine strain and the Brazilian 2012 H3N8 EIV; and (c) to access the inter and intra-host EIV genetic variation that might have caused the impact of this outbreak.
Materials and methodsSamplesNasal swabs were collected in triplicates from all the 32 horses that were in the hospital barns. The nasal swabs were transported in minimum essential medium (Gibco, Carlsbad, CA, USA), with penicillin (200U/mL) and streptomycin (200μg/mL) (Gibco, Carlsbad, CA, USA). Transtracheal washes were collected along with the nasal swabs, from eight horses that presented nasal discharge, cough and fever. The nasal swabs and transtracheal washes were kept at −80°C until use. A panel of 64 serum samples was collected from the horses: 32 from the first day on which they showed respiratory clinical signs; and 32 on the 15th day thereafter. The serum samples collected were stored at −20°C until use.
Influenza A virus screeningThe Directigen™ EZ FluA & B test kit (BD, Sparks, MD, USA) was used on nasal swabs from the first 13 horses that were suspected of having EI. This assay targets the influenza A virus nucleoprotein and was performed in accordance with the manufacturer's instructions.
Virus isolationVirus isolation was undertaken in 10-day-old specific pathogen-free (SPF) embryonated chicken eggs by means of the allantoic fluid route. The SPF eggs were kindly donated by Laboratório Biovet, located in Brazil. All horses that were positive for EIV by means of Directigen™ EZ FluA & B test kit (BD, Sparks, MD, USA) or amplification of M gene27,28 had nasal swabs (or transtracheal wash, in case it was done) used in virus isolation. Virus isolation (maximum of two passages) was confirmed by polymerase chain reaction (RT-PCR), targeting the M gene of influenza A viruses,27 under the conditions previously described.28
Amplification of M, HA and NA genes and DNA sequencingTotal RNA was extracted from 300μL of allantoic fluid of each sample using the Purelink® RNA mini kit (Ambion, Carlsbad, CA, USA) and cDNA was synthesized using 2.5μM of Uni12 primer (5′-NNAGCRAAAGCAGGNNN-3′, modified from Hoffmann et al., 2001) and SuperScript™ III (Life Technologies, Carlsbad, CA, USA). The RNA extraction and cDNA synthesis were done as per the manufacturer's instructions.
All nasal swabs were tested by means of RT-PCR targeting the M gene of influenza A viruses,27 under conditions described previously.28 Amplification of the complete hemagglutinin (HA) (1762 nt) and neuraminidase (NA) (1413 nt) genes from the isolates was undertaken using modified primers originally designed by Gildea et al.29 and Hoffmann et al.30 (Table S1) and Platinum™ Taq DNA Polymerase High Fidelity (Life Technologies, Carlsbad, CA, USA), as per the manufacturer's instructions. HA and NA amplicons were purified from agarose gels using Illustra™ GFX PCR DNA and gel band purification kit (GE Healthcare, Little Chalfont, Buckinghamshire, UK), as per the manufacturer's instructions. The sequencing reaction was carried out using the BigDye® Terminator v.3.1 Cycle Sequencing kit (Applied Biosystems, Carlsbad, CA, USA), in accordance with the manufacturer's instructions, in the Applied Biosystems™ 3500 Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). All fragments were amplified and sequenced three times, in order to increase confidence in the analysis.
Molecular cloningIn order to evaluate intra-host EIV genetic variation (quasispecies), a segment of HA gene was cloned and sequenced from four isolates. The HA RT-PCR products from the primers HA1F and HA466R29 were purified using ExoSAP-It® (USB, Cleveland, OH, USA) and were inserted into the pTZ57R/T vector of the Thermo Scientific™ InsTAclone PCR cloning kit (Thermo Fisher Scientific Inc., Vilnius, Lithuania) in Escherichia coli (JM109). Plasmids were extracted using Nucleospin® Plasmid (Macherey-Nagel, Düren, Germany) and were sequenced with M13/pUC primers (Thermo Fisher Scientific Inc., Vilnius, Lithuania) as described above.
Phylogenetic analysisThe confidence levels of all sequences were evaluated using the FinchTV™ software. Nucleotide sequences were applied using the Basic Local Alignment Search Tool (BLASTn), available in GenBank, National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov/BLAST/), to search for homologous sequences. EIV sequences from the EpiFlu™ database of the Global Initiative on Sharing All Influenza Data (GISAID) were also used in the analysis. The accession numbers in the GenBank and GISAID databases are provided as Supplementary data, in Tables S2 and S3. Sequences were aligned using Muscle,31 and maximum-likelihood amino acid trees were built using 1000 bootstrap replicates,32 through MEGA 7.33 The mean evolutionary diversity and distances for complete HA sequences were estimated using MEGA 7.33 HA nt sequences of eight isolates were compared in order to evaluate inter-host EIV genetic variation.
Evolutionary analysisA molecular clock test on 77 EIV HA1 nucleotide sequences representing the five clades was performed using the maximum likelihood method, with the Hasegawa–Kishino–Yano (HKY) model34 with discrete gamma distribution, in MEGA 7.33
The nucleotide substitution rates and phylogenetic relationships were assessed through Bayesian Markov Chain Monte Carlo (MCMC) analysis,35 using BEAST v.1.7.536 with the HKY model with four categories of gamma-distributed rate variation among sites.34 The strict and relaxed (lognormal and exponential) molecular clocks were calibrated using the strain dates. The effective sample size (ESS) was accessed by means of Tracer v.1.6.037 and the final MCC clock-tree was viewed using FigTree 1.4.2.38
Anti-influenza A virus antibodiesA commercial enzyme-linked immunosorbent assay (ELISA; FlockChek® Avian Multiscreen Antibody Test Kit, IDEXX Laboratories Inc., Westbrook, ME, USA) that had been developed to detect antibodies against the highly conserved viral nucleoprotein (NP) of the influenza A virus was used with the horse serum samples. The test consists in a competitive ELISA thus a higher concentration of specific antibodies will result in lower test result. Samples showing a ratio of the serum sample optical density (OD) to the negative control OD provided by the assay (S/N)≤0.60 were considered positive; while S/N>0.60 was considered negative for the presence of anti-influenza antibodies.
ResultsEI outbreak descriptionThe Veterinary School Hospital of the School of Veterinary Medicine and Animal Science, University of São Paulo, São Paulo, SP, Brazil, experienced an EIV outbreak in vaccinated and unvaccinated horses from September 16 to September 25, 2015. Twenty-six of the 32 horses housed at the hospital at the time of the outbreak (29 adult horses, two foals and one pony) displayed at least one clinical sign of EI within an eight-day period, including nasal discharge, dry cough and sneezing, along with fever, inappetence and mucosal congestion.
Among the 32 horses, ten horses had been immunized with a vaccine containing an inactivated A/equine/Kentucky/97 strain, and two had been immunized with the inactivated A/equine/South Africa/4/2003 and A/equine/Kentucky/94 strains. Three horses were immunized but the name of the vaccine and/or strain was not available. The two foals were from vaccinated mares but there was no full vaccination history available. For the remaining horses, the anti-EIV vaccination history was unknown.
Influenza A virus screening and isolationThe initial screening on the 13 horses that first showed respiratory clinical signs was carried out using the Directigen™ EZ FluA & B test kit (BD, Sparks, MD, USA), on nasal swabs. There were six positive results and one inconclusive result. The matrix (M) gene of the influenza A virus was identified by means of RT-PCR on 23 nasal swabs out of 32 that were tested. For the eight horses from which transtracheal washes were collected along with the nasal swabs, both samples were analyzed by means of RT-PCR for the M gene. Among these, seven swabs and three transtracheal washes showed positive results. The transtracheal washes (positive or negative) from positive horses (positive at nasal swab sampling) were used at virus isolation. Twelve EIVs were isolated from the 23 horses that were found to be positive through RT-PCR. Six of the isolates were from transtracheal washes and five were from nasal swabs. Twelve of the 15 vaccinated horses were infected with EIV, ten with clinical signs. One of the horses that showed clinical signs had no virus detected but had seroconversion at ELISA. Among the vaccinated horses that had EIV, two received the OIE recommended strain, eight the A/equine/Kentucky/97 and two with no description of which EIV strain received.
Anti-influenza A virus antibodiesThe ELISA results among the horses that were positive for EIVs were S/N 1.0918 (95% CI: 0.8612; 1.3225) at the first sampling and S/N 0.2620 (95% CI: 0.1607; 0.3633) on the 15th day thereafter (Table S4). Increased levels of influenza A antibodies were also seen with paired serum samples from the horses that virus was not detected or isolated from samples: S/N 0.6626 (0.3437; 0.9815) on day 1 and S/N 0.3615 (0.0995; 0.6235) on day 15. Nineteen horses seroconverted, nine were positive at both sampling and four remained negative. Twenty seven horses were considered infected due to virus detection in nasal swab or transtracheal wash, or to seroconversion at ELISA (negative-positive). The description of the samples, EIV screening, virus isolation, ELISA and vaccination status are presented in Table S4.
HA and NA gene analysesHA sequences were obtained from ten of the isolates. Six nucleotide (nt) sequences comprised full codons of the hemagglutinin gene (1698 nt), which included the signal peptide (nt 1–45) (SP/8, SP/10, SP/11, SP/12, SP/15 and SP/32), and four were from partial segments of the uncleaved protein: nucleotides 1 to 1016 (SP/4), 27 to 1698 (SP/25), 1 to 1165 (SP/30) and 1 to 437 (SP/18). Five neuraminidase sequences were obtained (SP/8, SP/10, SP/12, SP/15 and SP/25) comprising nt 53–1366, and no nucleotide differences were shown. For one representative strain (SP/8), NA was resequenced to achieve better NA gene coverage, from which nt 28–1395 were obtained. The HA and NA sequences of the EIV São Paulo/2015 strains were deposited in GenBank (KX954135–KX954164).
Nine amino acid substitutions were found in the HA1 sequences of São Paulo/2015 isolates, in comparison with the World Organisation for Animal Health (OIE)-recommended vaccine strain A/equine/South Africa/4/2003. Four of these substitutions were not present in the Brazilian 2012 outbreak strain (N3S, T121S, G142R and K304E) (Table 1).
Amino acid substitutions in complete HA sequences among Brazilian-2015 isolates from the present study (*), North and South American-2012 strains and the OIE-recommended Florida Clade 1 A/equine/South Africa/4/2003 vaccine strain. Numbers below sites E and A are the amino acid positions.
| Strain | Site E | Site A | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 | 7 | 62 | 104 | 121 | 138 | 142 | 223 | 304 | |
| A/equine/South Africa/4/2003 | N | G | R | D | T | A | G | V | K |
| A/equine/Sao Paulo/4.FMVZ/2015* | S | D | K | N | T | S | R | I | K |
| A/equine/Sao Paulo/8.FMVZ/2015* | S | D | K | N | T | S | R | I | K |
| A/equine/Sao Paulo/10.FMVZ/2015* | S | D | K | N | T | S | R | I | E |
| A/equine/Sao Paulo/11.FMVZ/2015* | S | D | K | N | T | S | R | I | E |
| A/equine/Sao Paulo/12.FMVZ/2015* | S | D | K | N | S | S | R | I | K |
| A/equine/Sao Paulo/15.FMVZ/2015* | S | D | K | N | S | S | R | I | K |
| A/equine/Sao Paulo/18.FMVZ/2015* | S | G/D | K | N | S | – | – | – | – |
| A/equine/Sao Paulo/25.FMVZ/2015* | S | D | K | N | S | S | R | I | K |
| A/equine/Sao Paulo/30.FMVZ/2015* | S | D | K | N | T | S | R | I | K |
| A/equine/Sao Paulo/32.FMVZ/2015* | S | D | K | N | S | S | R | I | K |
| A/equine/Rio Grande do Sul/2012 | N | D | K | N | T | S | G | I | K |
| A/equine/Dubai/1/2012 | N | D | K | N | T | S | G | I | K |
| A/equine/Sao Paulo/6/1963 | N | D | R | D | M | A | G | V | K |
A/equine/Dubai/1/2012 representing KF026412.1, KF026413.1, KF026407.1, KF026408.1, KF026409.1 and KJ372713.1.
A/equine/Sao Paulo/6/1963 representing CY032397.1
(–): sequence not available.
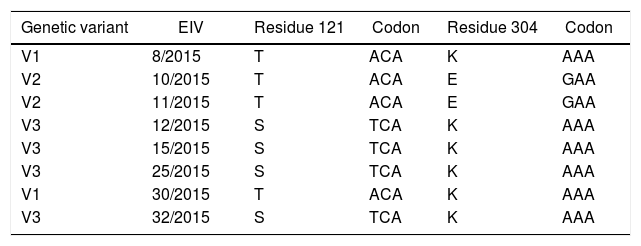
Three genetic variants were identified from the outbreak (variant 1, V1; variant 2, V2; and variant 3, V3) with single substitutions in residues 121 and 304 (T-K/T-E and S-K) at HA1 (Table 2). The sequence similarities between variants were 99.8% (V2/V3) and 99.9% (V1/V2 and V3) for nucleotides and 99.6% (V2/V3) and 99.8% (V1/V2 and V3) for amino acids.
Amino acid and nucleotide substitutions in the hemagglutinin gene (HA1 subunit) of eight equine influenza viruses isolated from an outbreak in São Paulo, SP, Brazil, 2015.
| Genetic variant | EIV | Residue 121 | Codon | Residue 304 | Codon |
|---|---|---|---|---|---|
| V1 | 8/2015 | T | ACA | K | AAA |
| V2 | 10/2015 | T | ACA | E | GAA |
| V2 | 11/2015 | T | ACA | E | GAA |
| V3 | 12/2015 | S | TCA | K | AAA |
| V3 | 15/2015 | S | TCA | K | AAA |
| V3 | 25/2015 | S | TCA | K | AAA |
| V1 | 30/2015 | T | ACA | K | AAA |
| V3 | 32/2015 | S | TCA | K | AAA |
The HA and NA amino acid maximum composite likelihood phylogenetic trees showed similar topologies and presented five groups: Florida Clade 1, Florida Clade 2, Eurasian, Kentucky and Pre-divergence (Figs. 1 and 2), showing eight HA1 sequences and one representative NA sequence from samples from this study. The viruses isolated in the present study belonged to the H3N8 subtype Florida Clade 1.
 of H3N8 EIVs available in GenBank and GISAID EpiFlu™ for the Florida Clade 1 (FC1), Florida Clade 2 (FC2), Kentucky, Eurasian and Pre-divergent lineages. The viruses characterized in the present study are indicated by diamonds (♦).")
Maximum composite likelihood amino acid tree, using Poisson model with 1000 bootstrap replicates of 77 HA1 subunit sequences (nt 1–987) of H3N8 EIVs available in GenBank and GISAID EpiFlu™ for the Florida Clade 1 (FC1), Florida Clade 2 (FC2), Kentucky, Eurasian and Pre-divergent lineages. The viruses characterized in the present study are indicated by diamonds (♦).
 of H3N8 EIVs available in GenBank and GISAID EpiFlu™ for the Florida Clade 1 (FC1), Florida Clade 2 (FC2), Kentucky, Eurasian and Pre-divergent lineages. The virus characterized in the present study is indicated by a diamond (♦).")
Maximum composite likelihood amino acid tree, using Poisson model with 1000 bootstrap replicates of 65 NA sequences (nt 28–1395) of H3N8 EIVs available in GenBank and GISAID EpiFlu™ for the Florida Clade 1 (FC1), Florida Clade 2 (FC2), Kentucky, Eurasian and Pre-divergent lineages. The virus characterized in the present study is indicated by a diamond (♦).
The hydrophobicity of HA1 changed in residues 62, 138, 142 of V1, V2 and V3 and 304 of V2, in comparison with A/equine/Rio Grande do Sul/1/2012 and A/equine/South Africa/4/2003 (Fig. 3). NA substitutions also changed the hydrophobicity profile. São Paulo/15 NA sequences had 39 nt changes and there were 11 aa changes in relation to A/equine/Ohio1/03 (FC1): one in the transmembrane helix (V35A) and ten in the head domain. The numbering of active sites followed the N2 numbering pattern.9 Five amino acid substitutions were found in SP-2015 NA but not in A/equine/Rio Grande do Sul/1/2012: L36P, R40G, R171K, E411G and H450Y. The 2012 and 2015 Brazilian strains shared the substitutions V35A, N205S, R260K, E271G, S337N and G416E.
 of the OIE-recommended strains, São Paulo/2015 isolates and A/equine/Rio Grande do Sul/1/2012 isolate. This profile was built using BioEdit v.7.2.5.39")
Eisenberg scale mean hydrophobicity profile for the equine influenza virus HA1 and partial NA amino acid sequences (465 aa) of the OIE-recommended strains, São Paulo/2015 isolates and A/equine/Rio Grande do Sul/1/2012 isolate. This profile was built using BioEdit v.7.2.5.39
The mean evolutionary diversity for the 157 complete HA sequences representing the five clades from 1963 to 2015 was 0.034 (SE 0.004) and 0.0019 (SE 0.0012) for the Brazilian 2015 lineages. In addition, the amino acid distance was calculated using the Poisson correction model40 in MEGA 733 (Table 3).
The molecular clock tests on 77 EIV HA1 sequences, representing the five clades, showed that the evolutionary rates among sequences were not equal. In both of the relaxed molecular clocks (lognormal and exponential), the ESS was above the minimum threshold of 200, which means that the sequences had good posterior representation and were uncorrelated. The lognormal clock better represented the evolutionary analysis than did the exponential clock, since it had lower standard deviation (SD): lognormal SD: 12.1418; exponential SD: 13.0462. Thus, the lognormal molecular clock was used for the MCMC analysis, with the HKY model and calibration using strain dates from BEAST v.1.7.536 (Fig. 4). The lineages grouped into two different clades: Clade 1 for V1 and V2; and Clade 2 for V3. The estimation for the time that has elapsed since the most recent common ancestor (TMRCA) indicated that the Brazilian-2015 strains are significantly younger than the root height and Florida Clade 1.
 and strain dates from BEAST v.1.7.5.36 The viruses characterized in the present study are indicated by diamonds (♦).")
MCMC evolutionary analysis on the HA gene of the EIV H3N8, using a lognormal molecular clock, HKY model and calibration with 77 HA1 sequences (nt 1–987) and strain dates from BEAST v.1.7.5.36 The viruses characterized in the present study are indicated by diamonds (♦).
HA sequences (1–437 nt of the gene) comprising the signal peptide and the first 130 amino acids of HA1 (nt 1–392) were cloned from three isolates (A/equine/São Paulo/8.FMVZ/2015, A/equine/São Paulo/10.FMVZ/2015, A/equine/São Paulo/15.FMVZ/2015) and one nasal swab (A/equine/São Paulo/18.FMVZ/2015). Four clones from each strain were sequenced and the amino acid and nucleotide mean diversities were estimated. The number of nucleotide and amino acid substitutions per site, relating to the mean diversity40 among the clonal populations of the four horses, was 0.0055 and 0.015, with standard error estimations of 0.001 and 0.004, respectively. The amino acid clone diversity was obtained using the Poisson model,41 and the nucleotide clone diversity was assessed using the maximum composite likelihood, both with gamma distributed rates and 1000 bootstrap replicates in MEGA 7.33
The descriptions of amino acid substitutions in partial HA1 clones from the strains A/equine/São Paulo/8.FMVZ/2015, A/equine/São Paulo/10.FMVZ/2015, A/equine/São Paulo/15.FMVZ/2015, A/equine/São Paulo/18.FMVZ/2015, A/equine/São Paulo/6/1963, A/equine/São Paulo/1/1969 and A/equine/South Africa/4/2003 are presented in Table S5.
DiscussionTwelve EIVs (H3N8) were isolated from horses during the outbreak, which ten were sequenced and three genetic variants (V1-3) were identified. The difference between V1/V2 and V3 consists of substitution in the residues 121 and 304, been V1 T121/K304, V2 T121/E304 and V3 S121/K304. Up to the time of this study, the residue S121 was unique to V3 and T121/E304 to V2 Brazilian 2015 strains, except by A/equine/Romania/1980 that have the V2 pattern. During the outbreak, this substitution first appeared in samples collected between September 19 and September 22, but was not seen in samples collected between September 14 and September 16, thus suggesting that it appeared during the outbreak.
Several equine flu outbreaks occurred around the world in 2010–2012 and were related to the FC1 and FC2 sublineages.11–16 The Brazilian-2015 isolates were similar to the Dubai, Uruguay, Argentina, Chile and Kentucky strains from 2012. The Brazilian-2015 strains were placed into two clusters in a major cluster that contains the FC1 EIV strains from 2011 and 2012. The short distances between the variants of the present study shown in each branch would be expected in strains from same outbreaks. The MCMC analysis indicated that the Brazilian 2015 strains and the 2012 strains from Dubai, USA and Brazil have the same ancestor. These variants clustered into two clades in the MCC tree: Clades 1 and 2.
Substitutions at HA antigenic sites can interfere with the antibody binding response.42 The EIV isolates in the present study had two amino acid substitutions at site A (residues 138 and 142) and one at site E (residue 62) when compared to A/equine/South Africa/4/2003. Site A showed decreased hydrophobicity and is situated in an external portion at the HA head.16,42 The two substitutions at site A (aa 138 and 142), which were seen in all the Brazilian EIV strains from 2015, showed that the combination of these two resulted in greater change when compared to the single substitution (A138S) found in A/equine/Rio Grande do Sul/2012 and A/equine/Dubai/1/2012 (Fig. 3), thus could affect antibody binding. Part of the lower hydrophobicity was due to the 138 substitution, which occurred in FC1-2012 strains but did not result in great antigenic changes.16 The substitution G142R was unique to V1, V2 and V3 Brazilian-2015 strains and was not found in other EIVs from public databases, thus suggesting that this substitution might have happened before or during the outbreak.
The substitutions R62K (site E), D104N and A138S (site A)6 and V223I were previously described in FC1 strains.14 The substitutions V78A and N159S that differentiate FC1 from FC2 remained.8,14 As noticed by Legrand et al.,14 the seven substitutions in residues G7D, R62K, V78A, D104N, A138S, N159S and V223I have apparently remained unchanged since 2008.
The nucleotide and amino acid diversity among the clones show that there were occurrences of quasispecies during the outbreak. This supports the hypothesis of variant evolution during outbreaks. A transmission network and evidence of genetic variants were previously studied in a single EIV outbreak.43
The catalytic function of the NA enzyme is related to the residues R-116, D-149, R-150, R-223, E-275, R-291, R-368 and Y-402,44 which were conserved in São Paulo-2015 strains. Epitopes at the NA head involving the residues 150, 199, 344–346, 367, 399 and 40045 were conserved among the EIVs analyzed, with three exceptions: the changes Q199H on A/equine/Kentucky/2/12 and A/equine/Kentucky/3/12 and R150Q on A/equine/Spain/1/07.
The ELISA used in this study has an epitope-blocking format and this format allows to be used for anti-influenza antibody detection in several species.46–48 Anti-NP antibodies were detected in nine horses in the first collection while in the second collection 28 samples were positive demonstrating serum-conversion after virus infection. Two horses that showed no clinical signs and were negative in EIV screening, seroconverted, confirming that they were infected during the outbreak.
The EIV variants isolated from this single outbreak suggest that a quasispecies evolutionary pattern was present. The substitutions (residues 138 and 142) found in HA1 might have affected antibody binding, especially to the horses that had been vaccinated with the OIE-recommended strain, A/equine/South Africa/4/2003. Also, the use of an out of date vaccine strain or updated vaccines without the production of protective antibody titers might be the major contributing factors on virus dissemination during this outbreak.
Ethical approvalThis study was approved by the Ethics Committee on Animal Use of the School of Veterinary Medicine and Animal Science of the University of São Paulo (no. 4480120314).
Conflicts of interestThe authors declare no conflicts of interest.
This work was financially supported by FAPESP-Brazil [2011/03234-7]; CNPq-Brazil [141677/2014-7] and CAPES/PROEX-Brazil [2327]. We would like to thank the Laboratório Biovet, located in Brazil, for the donation of the SPF embryonated chicken eggs used in this study. We gratefully acknowledge the contribution of the authors of the originating and submitting laboratories of the sequences in the EpiFlu™ database of GISAID on which this research is based. The list is detailed in Supplementary data shown in Tables S2 and S3.
The following are the supplementary data to this article: