


The soil represents the main source of novel biocatalysts and biomolecules of industrial relevance. We searched for hydrolases in silico in four shotgun metagenomes (4,079,223 sequences) obtained in a 13-year field trial carried out in southern Brazil, under the no-tillage (NT), or conventional tillage (CT) managements, with crop succession (CS, soybean/wheat), or crop rotation (CR, soybean/maize/wheat/lupine/oat). We identified 42,631 hydrolases belonging to five classes by comparing with the KEGG database, and 44,928 sequences by comparing with the NCBI-NR database. The abundance followed the order: lipases>laccases>cellulases>proteases>amylases>pectinases. Statistically significant differences were attributed to the tillage system, with the NT showing about five times more hydrolases than the CT system. The outstanding differences can be attributed to the management of crop residues, left on the soil surface in the NT, and mechanically broken and incorporated into the soil in the CT. Differences between the CS and the CR were slighter, 10% higher for the CS, but not statistically different. Most of the sequences belonged to fungi (Verticillium, and Colletotrichum for lipases and laccases, and Aspergillus for proteases), and to the archaea Sulfolobus acidocaldarius for amylases. Our results indicate that agricultural soils under conservative managements may represent a hotspot for bioprospection of hydrolases.
The soil is the richest habitat in microbial diversity, with estimates of containing about 10 billion micro-organisms per gram, encompassing thousands of species of bacteria, fungi and archaea,1 corresponding to approximately 1000Gbp of microbial genomes per gram of soil.2 With this microbial arsenal, the soil represents the main source for isolation of novel biocatalysts and other biomolecules of industrial relevance, such as antibiotics and enzymes. Soil micro-organisms and microbiomes have been studied in a variety of conditions, including undisturbed,3,4 agricultural,4–6 polluted or contaminated.7 Studies in several agroecosystems have allowed not only to get a better understanding of the microbial composition and functioning, but have also provided important tools for searching new molecules with potential biotechnological use.5,8,9
The intensification of land use for food production often results in negative impacts on soil quality, mostly caused by incorrect managements, including heavy use of fertilizers and pesticides, and practices that result in erosion and losses of soil organic matter. Therefore, soil and crop managements greatly influence the soil quality and may impact microbial community, affecting long-term sustainability.4,5,10–12 Soil management known as no-tillage (NT), in which sowing is performed directly into the crops’ residues, results in higher contents of soil organic matter, water content, and lower temperature oscillations, improving the conditions for micro-organisms acting on the biogeochemical processes, and allowing higher yields. On the contrary, the conventional tillage (CT) imposes constant soil stirring by plowing and disking, resulting in fast oxidation of soil organic matter, erosion, loss of water and biodiversity.4,5,10–12 Besides soil management, crop rotations including legumes and green manures are key for improving soil quality and reducing pests and diseases, increasing the sustainability of the cropping systems.4,5,10–12
The probability of finding micro-organisms with biotechnological potential for degradation of agrochemicals in agricultural soils is high.13 However, due to limitations of the classical methods for microbial isolation and growth, only about 1% of the soil microbial diversity is known.14,15 Nevertheless, the metagenomics approach independent of cultivation has revealed the hidden microbial potential, such as new microbial species, genes, and biomolecules.16
Hydrolases are among the most searched microbial molecules in soils; they compose a class of enzymes that catalyze the hydrolysis of covalent bonds, and have large industrial uses.17 Micro-organisms use hydrolases for degradation of natural organic polymers as source of energy; in addition, they are also involved in the metabolism of xenobiotics such as pesticides. New hydrolases with biotechnological potential, for example, lipases, amylases, proteases and cellulases, have been isolated by using different metagenomic strategies18,19; screening of enzymes in metagenomes is usually performed in silico, based on comparisons of gene sequences in databases.
The aim of this study was to identify hydrolases with potential industrial applications in an agricultural soil under different soil and crop managements in southern Brazil, through an in silico screening of shotgun DNA sequences obtained in four metagenomes.
Materials and methodsDescription of the field trial and soil samplingSoil metagenomes were obtained from soil samples of a 13-year-old experiment at the experimental station of Embrapa Soja, in Londrina, north of Paraná State, southern Brazil (23°11′S, 51°11′W, elevation of 620m). The soil is classified as Latossolo Vermelho Eutroférrico (Brazilian system), corresponding to Rhodic Eutrudox (US taxonomy). Soil chemical and physical properties and climatic conditions were given elsewhere.5 The treatments consisted of conventional tillage (CT) and no-tillage (NT), each under crop succession (CS) [soybean (Glycine max L. Merr.) in the summer and wheat (Triticum aestivum L.) in the winter], or crop rotation (CR) [soybean or maize (Zea mays L.) in the summer and wheat, lupine (Lupinus angustifolius L.) or oat (Avena strigosa Schreb.) in the winter]. The four treatments are designated as NTS, NTR, CTS and CTR. Other information such as cropping history, plot size, experimental design and replicates were given elsewhere.5 Soil samples were collected from the 0 to 10cm layer, in the rainy season, before sowing soybean (summer crop), three weeks after harvesting the winter crop, wheat. The great benefit of long-term experiments is that the effects of soil and crop managements reflect a large period, and the effects observed are cumulative, for example, as shown with samplings in different times of the year and different times of implementation performed in previous studies performed by our group.10,12
DNA extraction, shotgun sequencing and data processing have been described before.5,8 Shotgun sequencing resulted in about 1 million sequences for each treatment,5 and the datasets are deposited in the NCBI-SRA (National Center for Biotechnology Information- Sequence Read Archive) with the submission Accession Number SRA050780.
In silico screening based on nucleotide sequencesThe four soil metagenomes, totaling 4,079,223 sequences, were compared against sequences of hydrolases deposited at the NCBI-NR and KEGG (Kyoto Encyclopedia of Genes and Genomes) databases, and assigned according to the highest similarities. First, sets of data were created with the DNA sequences for each hydrolase (amylase, cellulase, laccase, lipase, pectinase and protease) from microorganisms (bacteria, archaea, fungi and virus) extracted from the NCBI database. The DNA sequences of each hydrolase were then compared with the sequences of the four soil metagenomes, by using the BlastX tool against the NCBI-NR and the KEGG databases.
Statistical analysisThe datasets were normalized by using the MG-RAST tools, as described before5; and analyzed with STAMP20 to identify differences in frequencies of DNA sequences coding for enzymes in the metagenomes, by comparing all combinations of treatments pairwise. Statistical significance was estimated with the G-test (w/Yates’) and Fisher's test at p≤0.05, using the Bonferroni's correction method, and DP:Asymptotic-CC for estimating the confidence interval.
Results and discussionLipasesSequences coding for lipases were the most abundant in the databases, with 23,038 sequences in the KEGG and 24,282 in the NCBI-NR databases. Lipases are abundant among animals, plants, and microorganisms, performing the breakdown of glycerol-ester bonds by hydrolysis21; they are common soil microbial enzymes, mainly produced by fungi.22
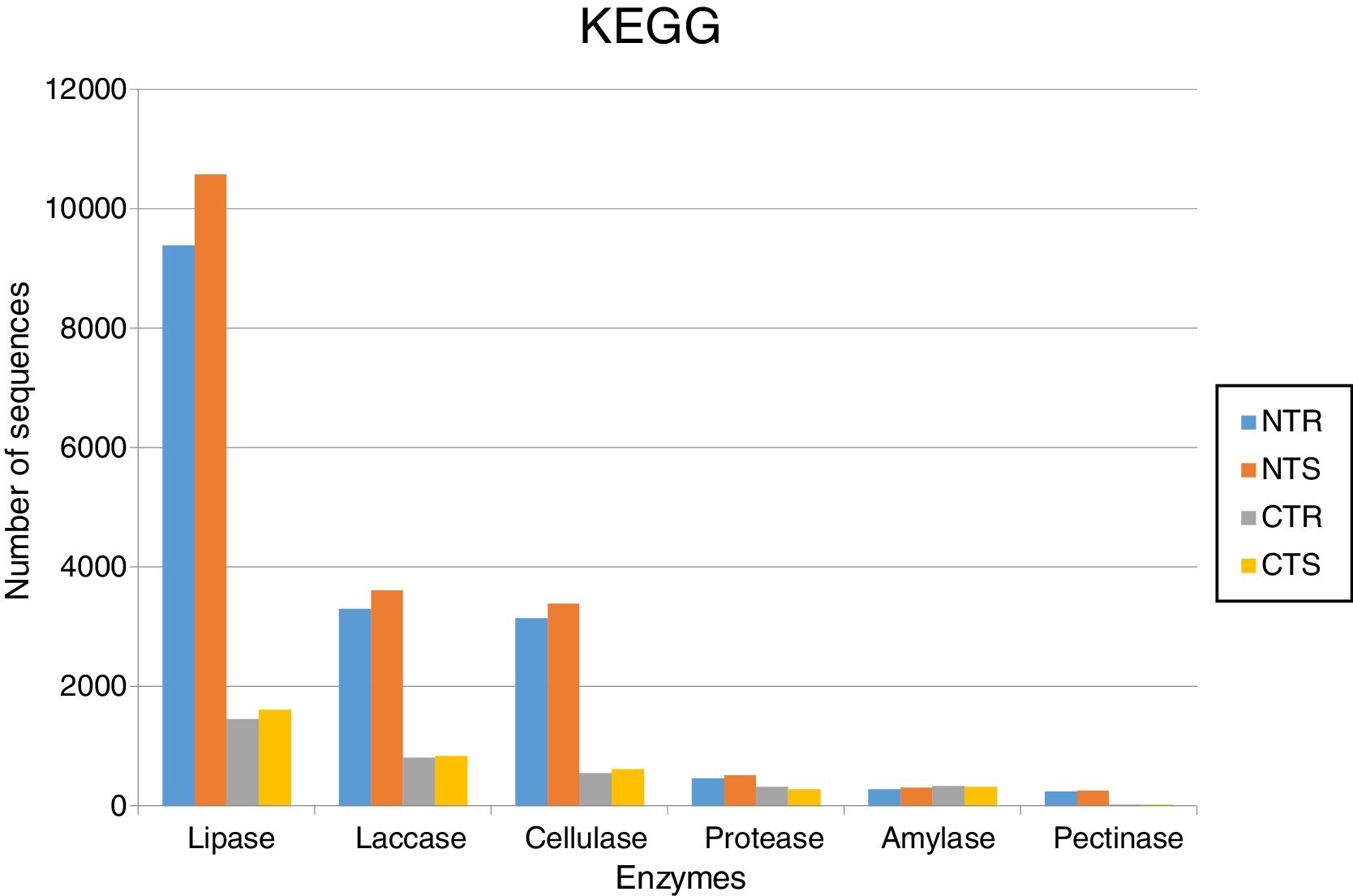
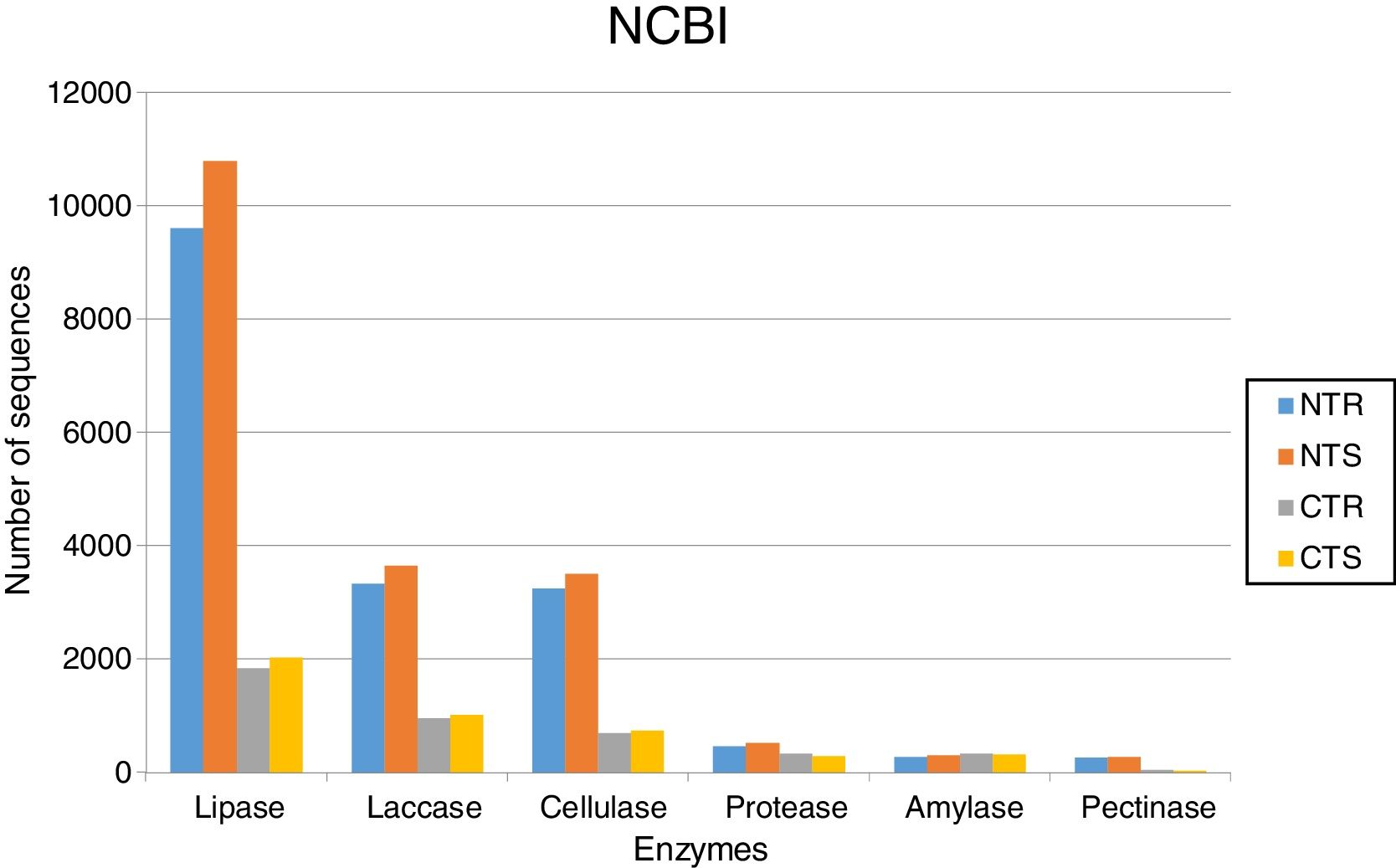
The soil management strongly affected the occurrence of lipases. Based on KEGG database, we identified 9392 sequences in the NTR (no-tillage with crop rotation), 10,582 in the NTS (no-tillage with crop succession), 1452 in the CTR (conventional tillage with crop rotation), and 1612 in the CTS (conventional tillage with crop succession) systems (Fig. 1). Based on NCBI-NR database, we found 9606 sequences in the NTR, 10,802 in the NTS, 1839 in the CTR, and 2035 in the CTS systems (Fig. 2).
 or conventional tillage (CT), with crop rotation (R) or crop succession (S).")
Abundance of DNA sequences coding for hydrolases identified with the BlastX tool showing homology with sequences deposited at KEGG database for each of the four metagenomes obtained from an oxisol in southern Brazil under 13-years of no-tillage (NT) or conventional tillage (CT), with crop rotation (R) or crop succession (S).
 or conventional tillage (CT), with crop rotation (R) or succession (S).")
Abundance of DNA sequences coding for hydrolases identified with the BlastX tool showing homology with sequences deposited at NCBI-NR database for each of the four metagenomes obtained from an oxisol in southern Brazil under 13-years of no-tillage (NT) or conventional tillage (CT), with crop rotation (R) or succession (S).
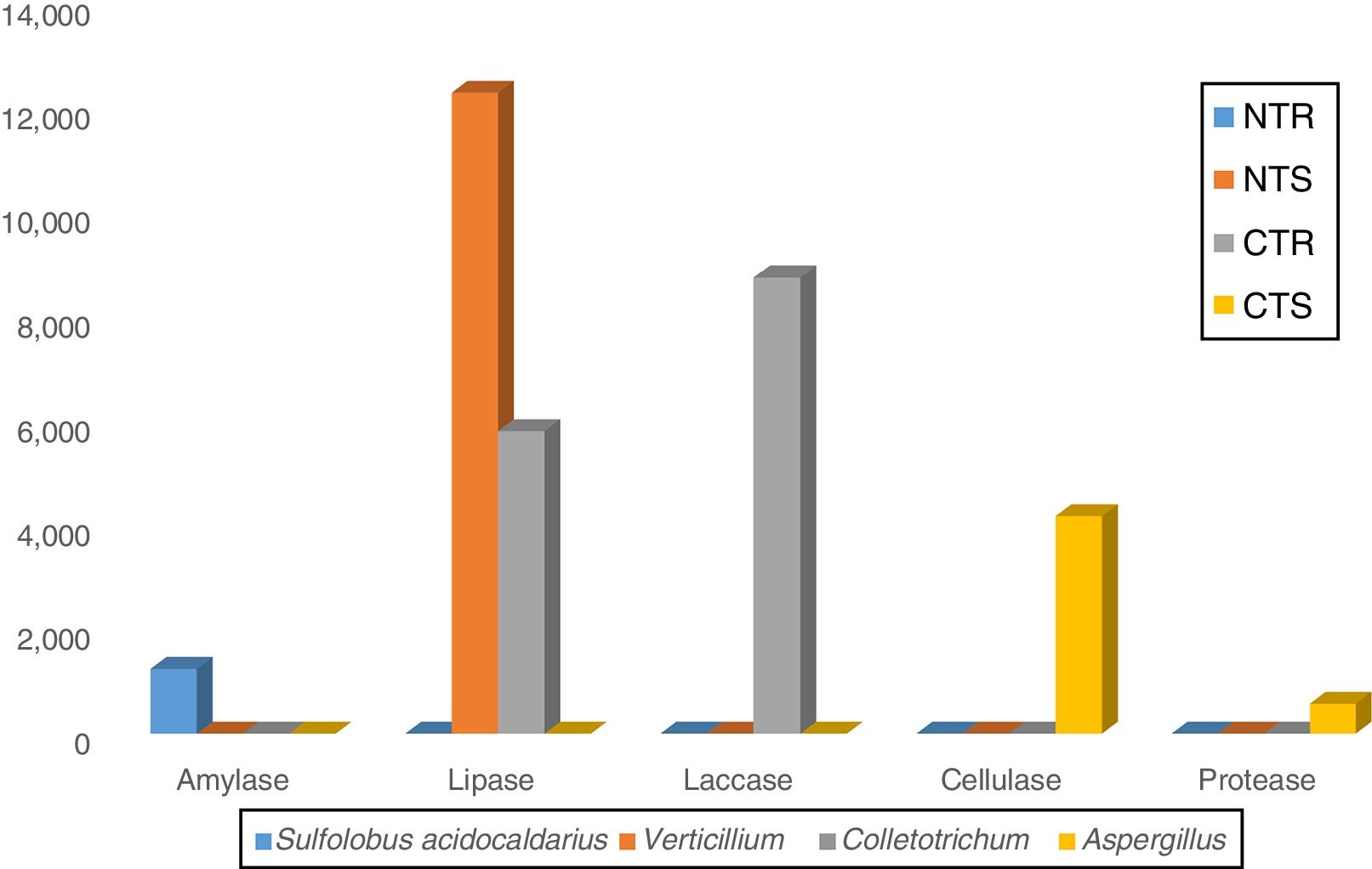
The majority of the lipases was associated to the fungal genera Verticillium (12,261 sequences) and Colletotrichum (5788 sequences) (Fig. 3), both belonging to the phylum Ascomycota. These are endophytic fungi known for their ability to synthesize and release lipases.23,24Colletotrichum has been recognized as the main producer of lipases in soil,22,23 and a study in a Peruvian soil revealed Verticillium as the producer of a new lipase with high hydrolytic activity.25

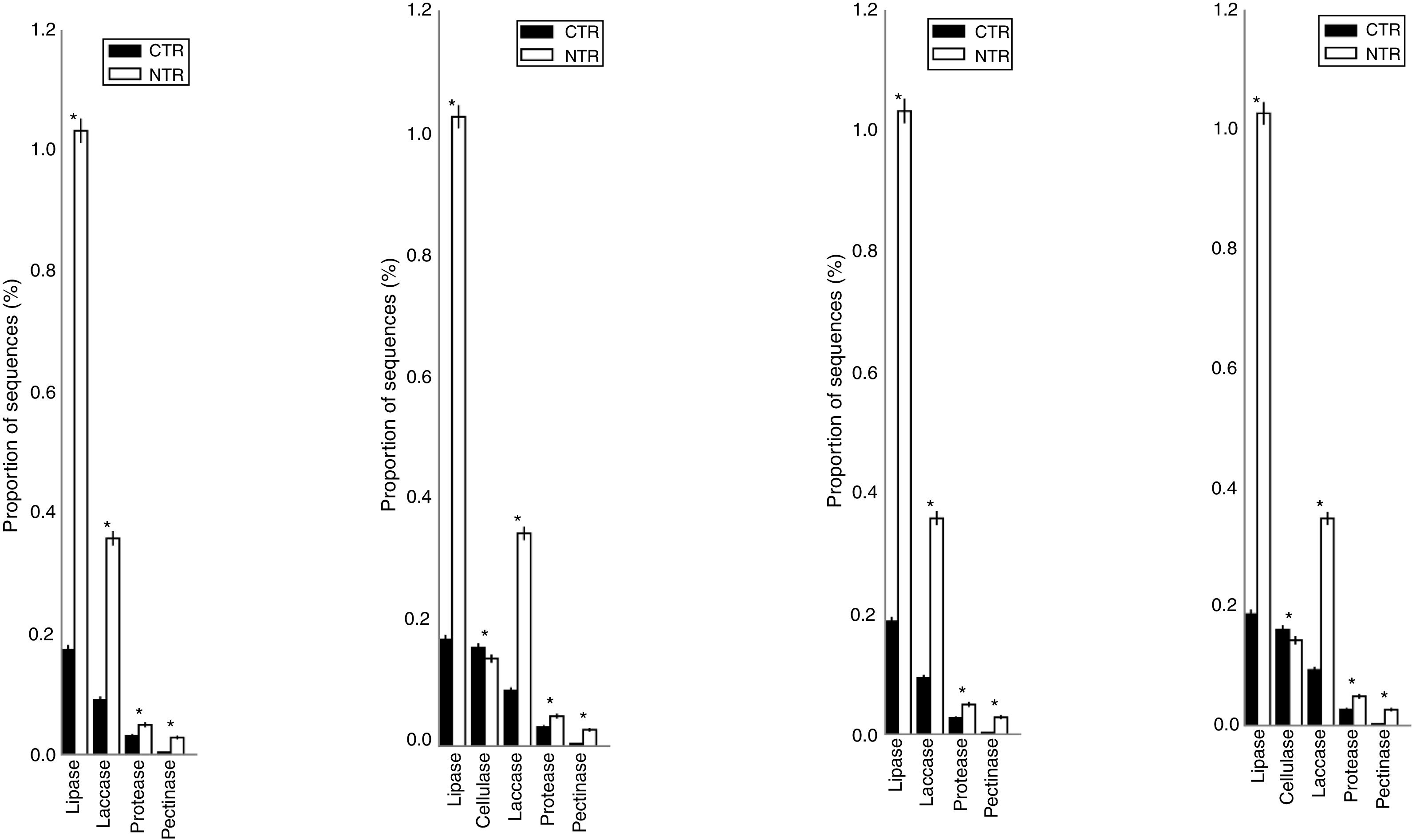
The statistically significant results for the pairwise comparison using STAMP are shown in Fig. 4. Differences on the occurrence of sequences coding for lipases were confirmed and mainly attributed to the tillage systems (p<0.05). On average, there were 550% more lipases in the NT than in the CT system; the CR presented 12% less sequences than the CS, but these differences were not statistically significant.
 and conventional tillage (CT) soil managements, under crop succession (S) or crop rotation (R) in a 13-year-old field experiment performed in an oxisol in southern Brazil. Only the hydrolases with statistical differences (p<0.05) are shown.")
Statistically significant differences for the frequencies of DNA sequences coding for hydrolases, evaluated with the STAMP software in the comparison between no-tillage (NT) and conventional tillage (CT) soil managements, under crop succession (S) or crop rotation (R) in a 13-year-old field experiment performed in an oxisol in southern Brazil. Only the hydrolases with statistical differences (p<0.05) are shown.
Lipids from oil seeds, such as soybeans, may stimulate the high abundance of lipases in agricultural soils. Both CT and NT soils have been cropped with soybean, but the main effect was attributed to the NT, which maintained the crop residues on the soil surface. Therefore, the NT favors not only the accumulation of organic matter, water retention, more favorable soil temperature conditions and microbial biomass,4,5,10–12 but also microbial diversity,4,5 and the higher abundance of lipases that we now report might contribute to the decomposition of the crop residues. In addition, lipases and organic matter may be related; indeed, Beyer et al.26 reported greater survival of Verticillium with higher organic matter content. The higher abundance of lipases under the NT is also important for the biogeochemical cycles, as lipids are sources of energy for soil microorganisms; they may also assist in environmental bioremediations (digesters, oil degradation, xenobiotics).27 Lipases are broadly used in industries, in processes such as the synthesis of biosurfactants, cosmetics, agrochemicals, food, detergents, paper, and oil processing.27,28 Due to their high economic value, the search for new lipases has grown fast, especially with studies of metagenomics; for example, Lee et al.29 analyzed 33,700 clones from a Korean forest soil and found novel lipase activities in six clones.
LaccasesLaccases were the second most abundant hydrolases in the four metagenomes of our study. Based on the KEGG database we found 8557 sequences, 3300 in the NTR, 3609 in the NTS, 807 in the CTR, and 841 in the CTS systems (Fig. 1). Similar numbers were found based on the comparison with the NCBI-NR database, with 8964 sequences, 3332 in the NTR, 3656 in the NTS, 960 in the CTR, and 1016 in the CTS systems (Fig. 2). The statistically more frequent occurrence of laccases in the NT system was confirmed (Fig. 3). The sequences were mostly assigned to the genus Colletotrichum (8724 sequences) (Fig. 3), which is known as a natural producer of laccases, property that has often been related to pathogenicity.30,31 Moreover, laccases are also very important for bioremediation, due to their ability to oxidize an array of compounds, such as pesticides, as well as lignin.32 However, one must consider that that the fungal data bases are limited in laccase sequences that have been annotated, and that other genera can be important for their production.
The biotechnological potential of laccases has led to their bioprospection in metagenomes in several environments. For example, Fang et al.33 reported a new bacterial laccase from a marine microbial metagenome in south China. Further studies based on in silico strategies found numerous new laccases in soil samples.34 Therefore, our study shows that soils under the NT management may represent a hotspot of laccases-coding genes to be prospected.
CellulasesCellulases comprise several enzymes with the ability to hydrolyze cellulose, with a variety of biotechnological applications, such as the production of second-generation ethanol from cellulosic materials, in paper and textile industries, and food processing.35,36 In our study, cellulases were the third most abundant hydrolases. Based on the KEGG database, we found 7702 cellulase sequences, 3145 in the NTR, 3389 in the NTS, 553 in the CTR, and 615 in the CTS systems (Fig. 1). The NCBI-NR database revealed 8204 cellulases, 3250 in the NTR, 3505 in the NTS, 700 in the CTR, and 749 in the CTS systems (Fig. 2). The majority of the sequences were assigned to the Aspergillus genus (4175 sequences) (Fig. 3), Ascomycota phylum. Fungi such as Aspergillus niger, Penicillium sp. and Fusarium oxysporum are reported as important cellulases producers.35–37 In our study, once more, the statistical difference was attributed to the soil management (NT×CT) (Fig. 4).
Soils under the NT system are considered more suppressive to pathogens than the CT, and we may hypothesize that the higher abundance of cellulases in the NT may help in the control of pests and diseases. Indeed, the ability of some microorganisms to control pathogens has often been associated with their ability to produce cellulases, for example, the purified enzymes of Trichoderma harzianum were able to inhibit conidial germination and germ tube elongation of the surviving spores.38 In addition, cellulases are also important in the C biogeochemical cycle, where cellulose represents an important source of energy and C for soil microorganisms.
ProteasesProteases are enzymes that catalyze the hydrolysis of proteins and are critical for microbial performance in all environments; in our study, DNA sequences coding for proteases were the fourth most abundant. The comparison with the KEGG and NCBI-NR databases identified 1567 and 1604 sequences, respectively. In the KEGG database, 459 sequences were found in the NTR, 510 sequences in the NTS, 318 in the CTR, and 280 in the CTS systems (Fig. 1). In the NCBI-NR database, 462 sequences were assigned to the NTR, 518 to the NTS, 330 to the CTR, and 294 to the CTS systems (Fig. 2). Once more, statistically higher frequency of proteases was found in the NT treatment, especially the NTS, while the lowest was detected in the CTS (Fig. 4). Crop residues represent the main source of nutrients for microbes, and most proteins in soils are of microbial origin, being important constituents of the soil N pools.39 It has been reported that higher availability of N and C—as usually found in the NT system10–12—implies in higher microbial proteolytic activity,40 and we may hypothesize that favoring the slow decomposition of crop residues, and increasing the N availability, the protein content would be higher, selecting proteolytic microorganisms, as indicated in our study. On the other hand, the CT speeds up the decomposition rate and losses of soil N pools, and consequently fewer organisms specialized in proteolysis remain. However, proteolytic activities are found even in soils containing very low pools of protein N, as reported for a Bacillus from a desert soil in India,41 and confirmed in our study in the treatment poorest on C and N, the CTS.
In all soil and crop management systems, the majority of the proteases were assigned to A. niger (575 sequences) (Fig. 3), but, again, we should comment that the databases are still quite limited in annotated fungal protease sequences. Several studies have shown major fungal capacity in the production of proteases.24,42 Fungi are biotechnologically interesting as proteases sources because of the easy growth, fast production and easy removal of the mycelium, resulting in lower costs.43 Devi et al.44 purified and characterized an alkaline protease produced by A. niger isolated from an Indian soil, showing high compatibility for use in commercial detergents; other proteases produced by A. niger have been described with applications in food, laundry, detergent, and pharmaceutical industries. Bioprospection of proteases has increased with the introduction of metagenomics approaches.45
AmylasesAmylases are extracellular hydrolytic enzymes broadly distributed in microbial, plant, and animal kingdoms, acting mainly on starch degradation,46 but can also hydrolyze amylose, amylopectin, cyclodextrins, glycogen, and dextrins.47 Amylases are classified into three types, according to the cleavage site: alpha-, beta- and gamma-amylases. Microbial production is well defined in Bacillus for the alpha-type,48 and has been reported in different soils, e.g., in mangrove orchards in India.49 Alpha-amylases are one of the most popular and important industrial amylases, with broad applications in food, textile and detergents sectors.49,50 Therefore, the search for amylases has been a goal in several studies, now with an emphasis on metagenomes.28,50,51
In our study, amylases were the fifth most abundant hydrolases among the metagenomes. The search in the KEGG database identified 1235 sequences, 303 in the NTS, 276 in the NTR, 336 in the CTR, and 320 in the CTS systems (Fig. 1). In the NCBI-NR database, we found 1247 amylase-coding sequences, 309 in the NTS, 278 sequences in the NTR, 339 in the CTR, and 321 in the CTS systems (Fig. 2). There were no statistical differences between the treatments concerning the frequencies of DNA sequences coding for amylases. The amylases were mainly related to Sulfolobus acidocaldarius, belonging to the Archaea domain (1247 sequences) (Fig. 3), which in our previous taxonomy study revealed to be most abundant in soils under the NT system.5 The production of amylases by Archaea has been reported in different environments,47,52 but is not as common as for Bacteria such as Bacillus. Therefore, the abundance of DNA sequences from Archaea coding for amylases in our study deserves further investigation, as they may represent novel types.
PectinasesPectinases are present in several microorganisms and plants, acting on pectin, which is the main component of plant cell walls.53 Pectinases have great economic value, being largely employed in the processing of textiles, coffee, plant fiber for juice production, and oil extraction.54,55
Pectinases-coding DNA sequences had the lowest abundance in our metagenomes. The search in KEGG database identified 532 sequences, 241 in the NTR, 251 in the NTS, 20 in the CTR, and 20 in the CTS systems (Fig. 1). In the NCBI-NR database, we found 627 sequences, 266 in the NTR, 283 in the NTS, 43 in the CTR, and 35 in the CTS system (Fig. 2).
We were not able to attribute the sequences to any specific micro-organism. However, the pectinolytic activity is common in several microorganisms, and Aspergillus has been frequently used for the commercial production.56,57 Agricultural soils usually receive pectin-rich crop residues,58 and the higher abundance of pectinases in the NT can be associated with the deposition of residues on the soil surface, favoring the microbial activity4,5,10–12 and diversity.4,5 The statistically higher frequency of DNA sequences coding for pectinase in the NT system (Fig. 4) could be explained by the higher content of soil organic matter. This hypothesis is supported by the study of Nisha and Kalaiselvi,59 who tested the pectinase activity with different sources of C and N, and verified that the synthesis was maximized at higher concentrations of C and N.
Metagenomics has been used to find new pectinases. For example, Sathya et al.60 found nine clones with pectinase activity from a forest soil of Southern Western Ghats, India, and Singh et al.61 found a thermo-stable pectinase in another Indian soil. Even though with relatively few sequences compared with the other hydrolases, the greater abundance of sequences coding for pectinases in the NT highlights the potential of finding new hydrolases of industrial importance in soils under conservative managements.
ConclusionsHydrolases have large biotechnological uses in industries of biosurfactants, detergents, cosmetics, agrochemicals, food, fibers, paper, textiles, laundry, oil, ethanol, and pharmaceutical. In our study, the search in four soil metagenomes resulted in the identification of 42,631 (KEGG database), and 44,928 (NCBI-NR) hydrolases, representing about 1% of the 4,079,223 sequences prospected. Based on the KEGG database, the abundances of DNA sequences coding for hydrolases followed the order: lipases (54.0%)>laccases (20.1%)>cellulases (18.1%)>proteases (3.7%)>amylases (2.9%)>pectinases (1.2%). Outstanding and statistically significant differences were attributed to tillage, with the NT showing almost five times more hydrolases than the CT system. Emphasis should be given to the higher abundance under the NT system, when compared to the CT, of lipases, laccases, cellulases, and pectinases, of 6.5-, 4.2-, 5.6-, and 12.1-fold, respectively (KEGG). One main inquire could be that in our study the sampling at the superficial layer (0–10cm), where NT is richer that the CT in crop residues would not represent the deeper layers. However, in a previous study in the same field experiment, we reported that differences between the NT and the CT in microbial biomass of C and N, as well as in C and N stocks were observed in all layers, up to 60cm.11 We may thus conclude that the differences between the NT and the CT in the pattern of residue decomposition resulted in profound differences in the abundance of hydrolases in a relatively short period, of 13 years. Differences between the crop succession and rotation were far lower—10% higher for the first one—but not statistically different. Therefore, increasing the number of crops did not impact on the abundance of DNA sequences coding for hydrolytic enzymes, probably because the diversity of plant residues was not enough to affect the microbial metabolic diversity. The results from our study showing higher abundance of hydrolases in the NT also correlate with higher microbial biomass and activities of several microbial enzymes under this system.62
Interestingly, microbial taxonomic5 and functional8 diversities in the same soil and treatments are in agreement with the results that we now report of abundance of hydrolases. Increases in fungal biomass in NT in comparison with the CT have been reported,63 as well as differences in the strategies of C utilization,64 and, in general, in our study, hydrolases sequences were attributed to fungi, Verticillium and Colletotrichum (lipases and laccases), and Aspergillus (proteases). Interestingly, for the amylases, sequences were attributed mainly to the archaea S. acidocaldarius; important roles of mesophilic and thermophilic Group I archaea in soil, more specifically in N-cycling (ammonia oxidizers) have been recently described,65 and now we extend their importance to the amylases. Previously, we demonstrated that fungi and archaea were benefited by the NT when compared to the CT system,5 and now we confirmed that these microorganisms are the main sources of hydrolases under NT. Finally, our results indicate that agricultural soils under conservative managements such as the NT may represent a hotspot for bioprospection of hydrolases for biotechnological and industrial applications. Further studies should now be conducted to try to characterize some hydrolases identified in our study.
Availability of data and materialsAll data and materials cited on the manuscript are freely available for the scientific community. Metagenome data are deposited at the NCBI-SRA with the Accession Number SRA050780.
FundingFinanced by CNPq-Universal (400468/2016-6), INCT-Plant-Growth Promoting Microorganisms for Agricultural Sustainability and Environmental Responsibility (CNPq 465133/2014-2 – Fundação Araucária-CAPES) and Embrapa (02.14.01.026.00.06.002).
Authors’ contributionMH and RCS initiated and designed the study.
MH and ATRV contributed with reagents/materials.
RCS and MEC performed the experiments.
RCS and MEC analyzed the data.
RCS and MH wrote the paper.
All authors read, contributed and approved the final manuscript.
Conflicts of interestAuthors declare no conflicts of interest or ethical problems.
RCS acknowledges a postdoctoral fellowship from CNPq. Financed by CNPq-Universal (400468/2016-6), INCT-Plant-Growth Promoting Microorganisms for Agricultural Sustainability and Environmental Responsibility (CNPq 465133/2014-4, Fundação Araucária-STI, CAPES), and Embrapa (02.14.01.026.00.06.002). MAN, ATRV and MH are also CNPq research fellows.