


Members of the Sphingomonas genus are often isolated from petroleum-contaminated soils due to their unique abilities to degrade polycyclic aromatic hydrocarbons (PAHs), which are important for in situ bioremediation. In this study, a combined phenotypic and genotypic approach using streptomycin-containing medium and Sphingomonas-specific PCR was developed to isolate and identify culturable Sphingomonas strains present in petroleum-contaminated soils in the Shenfu wastewater irrigation zone. Of the 15 soil samples examined, 12 soils yielded yellow streptomycin-resistant colonies. The largest number of yellow colony-forming units (CFUs) could reach 105CFUsg−1soil. The number of yellow CFUs had a significant positive correlation (p<0.05) with the ratio of PAHs to total petroleum hydrocarbons (TPH), indicating that Sphingomonas may play a key role in degrading the PAH fraction of the petroleum contaminants at this site. Sixty yellow colonies were selected randomly and analyzed by colony PCR using Sphingomonas-specific primers, out of which 48 isolates had PCR-positive signals. The 48 positive amplicons generated 8 distinct restriction fragment length polymorphism (RFLP) patterns, and 7 out of 8 phylotypes were identified as Sphingomonas by 16S rRNA gene sequencing of the representative strains. Within these 7 Sphingomonas strains, 6 strains were capable of using fluorene as the sole carbon source, while 2 strains were phenanthrene-degrading Sphingomonas. To the best of our knowledge, this is the first report to evaluate the relationship between PAHs contamination levels and culturable Sphingomonas in environmental samples.
Polycyclic aromatic hydrocarbons (PAHs) are considered hazardous due to their toxic, genotoxic, mutagenic and/or carcinogenic properties.1 Microbial degradation is a major process in the successful removal and elimination of PAHs from contaminated soils.2 The use of native or indigenous microbiota is of great interest for bioremediation of petroleum-polluted sites because they are often more useful and beneficial than commercial inocula that can be out-competed by indigenous microorganisms.3
Members of the genus Sphingomonas, which was proposed by Yabuuchi et al.,4 are frequently isolated from PAH-contaminated soils, suggesting that the Sphingomonas are probably key members of natural PAH-degrading microbial consortia.3,5,6 A number of studies demonstrated that Sphingomonas was one of the dominant populations in the microbial communities of PAH-contaminated soil and water samples.7–9
The Shenfu irrigation zone is located between Shenyang and Fushun city in Liaoning province and is the largest petroleum wastewater irrigation zone in China. The paddy fields in this region have been irrigated by oil-bearing wastewater discharged by the petroleum and coal-refining industry since the 1940s due to a lack of irrigation water. During the 1980–1990s, partial paddy fields were converted to upland in the up- and mid-stream. Mineral oil (especially PAHs) was the dominant contaminant in the Shenfu irrigation zone.10 Our previous study demonstrated that a large percentage of Sphingomonas species was present in the petroleum-contaminated soils of the Shenfu irrigation area using culture-independent methods,11 indicating a potential link between Sphingomonas and the biodegradation of petroleum compounds in the Shenfu irrigation zone. However, due to the lack of a reliable system for the specific detection and isolation of culturable Sphingomonas, the functional strains in the environmental samples were only found at random.
Vanbroekhoven et al.12 developed a streptomycin-based Sphingomonas growth medium that, together with the yellow pigmentation of Sphingomonas, facilitated the detection and isolation of culturable Sphingomonas from soils. However, this phenotypic detection method is not efficient or accurate because other bacterial taxa also produce yellow pigments and are resistant to streptomycin. A genotypic detection method that circumvents the major drawbacks inherent in the phenotypic detection method described above has been developed for the detection of specific groups. The 16S rRNA gene sequence offers ample possibilities to distinguish microorganisms of various genera and minimize false-positive reactions using genus-specific primers.13,14 PCR-based detection has proven to be useful for the identification of the genera Clostridium, Pseudonocardia, Saccharopolyspora, Nocardiopsis and Saccharothrix using genus-specific primers.14–16 In our previous study, a primer set targeting the Sphingomonas 16S rRNA gene was designed17; thus, we hypothesized that the use of genus-specific colony PCR combined with phenotypic identification would allow the specific detection of Sphingomonas isolates.
The specific aim of this study was to investigate the relationship between indigenous culturable Sphingomonas in soils from the Shenfu petroleum-wastewater irrigation zone and the contamination level by the direct and specific detection of culturable Sphingomonas from the soils. Additionally, the PAH-degrading capability of the isolated Sphingomonas was preliminarily characterized.
Materials and methodsSoil samplesSoil samples were collected from the Shenfu wastewater irrigation zone (41°50′46″–41°41′24″N, 123°44′43″–123°25′38″E), which is the biggest petroleum wastewater irrigation zone in China. The total length of the irrigation channel is approximately 70km, and 15 sampling sites with different relative geographical positions, irrigation histories and soil management types were selected from up- to down-stream. The 15 sampling sites included 11 paddy soils (soils A–K) and 4 upland soils (soils L, M, N and O) that were referred to as soils A–O in the following text. The physicochemical soil characteristics and the PAH concentrations in the soil were reported in our previous study.11 Typically, soil samples collected from the up- and mid-stream sites accumulated more petroleum contaminants compared with those from the down-stream sites.
Selective isolation and enumeration of putative SphingomonasThe indigenous putative culturable Sphingomonas were isolated and enumerated using a previously described method.12 The streptomycin-resistant yellow colonies that occurred on the plates were counted based on the formation of colony forming units (CFUs).
PCR and RFLP analysis of the Sphingomonas isolatesThe identity of the yellow colonies grown on the streptomycin plates was confirmed by genus-specific colony PCR. The PCR protocol was used with the SA429f/933r primer set developed in our previous study.17 The amplicons with the expected size were further analyzed by RFLP analysis using three endonucleases [HaeIII (GG′CC), HinfI (G′ANTC) and MspI (C′CGG)] at 37°C for 2h. The RFLP patterns were compared manually, and distinguishable RFLP patterns were considered to represent one phylotype.
Phylogenetic identification of Sphingomonas isolatesTwo representatives of each phylotype were selected for nearly full-length 16S rRNA gene sequencing for phylogenetic identification. PCR amplification of the 16S rRNA genes was performed with the forward primer 27F and the reverse primer 1492R.18 The obtained sequences were used for phylogenetic analysis as described previously.11
Measurement of PAH degradationTo determine the PAH-degrading capability of the isolated Sphingomonas strains, the pure strains were inoculated into mineral salts medium (MSM) containing a single PAH compound as the sole carbon and energy source. The MSM at pH 7.0–7.2 contained 2g K2HPO4, 0.5g KH2PO4, 0.5g NaCl, 0.5g NH4Cl, 0.2g MgSO4, 10mg CaCl2, and 1ml of trace element solution per liter of water. The composition of the trace element solution was 2gL−1 FeSO4·7H2O, 2gL−1 MnSO4·H2O, 0.5gL−1 Na2MoSO4·2H2O, 0.2gL−1 H3BO4, 0.4gL−1 CuSO4·5H2O, 0.5gL−1 ZnSO4, 0.2gL−1 NH4VO3, 0.5gL−1 CoCl2·6H2O, and 0.2gL−1 NiCl2·6H2O. Eight PAHs, including naphthalene (NA), acenaphtene (ACY), fluorene (FLO), phenanthrene (PHE), anthracene (ANT), fluoranthene (FLU), pyrene (PYR) and benzo[a]pyrene (BaP), were prepared at a concentration of 5gL−1 in acetone and filtered prior to analysis with a 0.22-μm Teflon syringe filter.
The PAH degradation experiment was performed by monitoring the disappearance of PAH in live versus killed controls. The experiment was conducted in triplicate in sterile dry 250ml Teflon-capped flasks. After all of the acetone in the PAH stocks was allowed to evaporate, 27ml of MSM broth and 3ml of inoculum (OD600=0.8–1.0) were added to each flask at a final concentration of 200μgml−1 of NA, ANT, FLU, PHE, ANT, and FLU, 100μgml−1 of PYR, and 50μgml−1 of BaP. Triplicate flasks were inoculated with live bacteria and 4% formalin-killed controls. The cultures were incubated in the dark on a rotary shaker (150rpm) at 30°C for 7 days, and then the flask contents were extracted for 30min with hexane (1:1, vol/vol). Flasks with PAHs but without the pure bacterial cultures were also run as a control. The extract was analyzed by high performance liquid chromatography (HPLC) using a Waters 2695 apparatus (Waters Inc., USA) equipped with an ultraviolet detector and a 250×4.6mm2 Inertsil ODS-P (Dikma Inc., USA) reverse phase C18 column. The HPLC analysis was performed using methanol:water (90:10, v/v) as the mobile phase at a flow rate of 0.7mlmin−1. PAH peaks were identified and integrated using the Masslynx software (Waters Corporation, Milford, MA).
Nucleotide sequence accession numbersThe nucleotide sequences have been deposited in the GenBank database under the following accession numbers: JF716058 to JF716065.
Statistical analysisAnalysis of all data was performed by ANOVA using SPSS version 16.0. Linear correlation coefficients were determined between different biological and biochemical parameters. P values below 0.05 were considered statistically significant.
Results and discussionEnumeration of putative Sphingomonas by plating on streptomycin agarMost Sphingomonas strains are characterized as yellow-pigmented, straight, rod-shaped bacteria. Additionally, resistance to streptomycin (Sm) is considered a common feature of Sphingomonas.12 Hence, yellow colonies on plates containing Sm were counted as putative culturable Sphingomonas. Previous real-time PCR analysis indicated that all 15 petroleum-contaminated soil samples gave positive signals for Sphingomonas.11 However, in this study only 12 soils yielded yellow colonies on the selective LB plates with Sm. The number of yellow CFUs ranged from 0.6×103 to 1.6×105CFUsg−1soil (Table 1). Provided that all of the yellow colonies belonged to the Sphingomonas genus, the counts of the Sphingomonas strains could only reach 105CFUsg−1soil, which was much less than the abundance (107–108 copiesg−1soil) obtained by our previous real-time PCR assay in the same tested soils.11 This discrepancy suggested that uncultured Sphingomonas accounted for a rather large proportion in the petroleum-contaminated soils, which was consistent with the results of Vanbroekhoven et al.12 and Leys et al.19 Although culturable Sphingomonas represent only a small percentage in the polluted soils, it is nevertheless important to obtain pure Sphingomonas cultures for functional and mechanical characterization due to their potential roles in bioremediation.
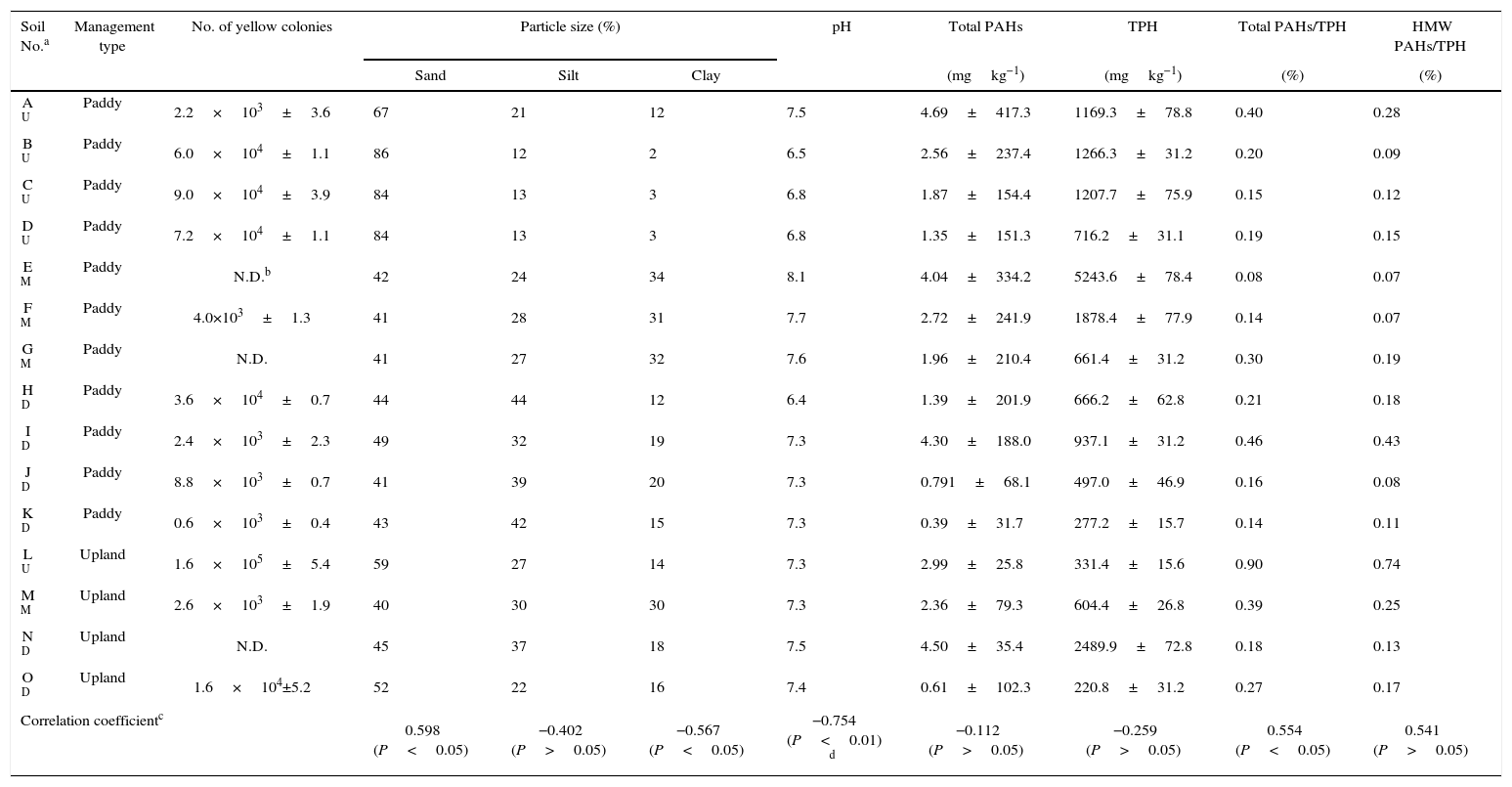
Total numbers of yellow streptomycin-resistant colonies in 12 soils from the Shenfu irrigation zone and the correlation between the number of yellow colonies and soil properties.
| Soil No.a | Management type | No. of yellow colonies | Particle size (%) | pH | Total PAHs | TPH | Total PAHs/TPH | HMW PAHs/TPH | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Sand | Silt | Clay | (mgkg−1) | (mgkg−1) | (%) | (%) | ||||
| A U | Paddy | 2.2×103±3.6 | 67 | 21 | 12 | 7.5 | 4.69±417.3 | 1169.3±78.8 | 0.40 | 0.28 |
| B U | Paddy | 6.0×104±1.1 | 86 | 12 | 2 | 6.5 | 2.56±237.4 | 1266.3±31.2 | 0.20 | 0.09 |
| C U | Paddy | 9.0×104±3.9 | 84 | 13 | 3 | 6.8 | 1.87±154.4 | 1207.7±75.9 | 0.15 | 0.12 |
| D U | Paddy | 7.2×104±1.1 | 84 | 13 | 3 | 6.8 | 1.35±151.3 | 716.2±31.1 | 0.19 | 0.15 |
| E M | Paddy | N.D.b | 42 | 24 | 34 | 8.1 | 4.04±334.2 | 5243.6±78.4 | 0.08 | 0.07 |
| F M | Paddy | 4.0×103±1.3 | 41 | 28 | 31 | 7.7 | 2.72±241.9 | 1878.4±77.9 | 0.14 | 0.07 |
| G M | Paddy | N.D. | 41 | 27 | 32 | 7.6 | 1.96±210.4 | 661.4±31.2 | 0.30 | 0.19 |
| H D | Paddy | 3.6×104±0.7 | 44 | 44 | 12 | 6.4 | 1.39±201.9 | 666.2±62.8 | 0.21 | 0.18 |
| I D | Paddy | 2.4×103±2.3 | 49 | 32 | 19 | 7.3 | 4.30±188.0 | 937.1±31.2 | 0.46 | 0.43 |
| J D | Paddy | 8.8×103±0.7 | 41 | 39 | 20 | 7.3 | 0.791±68.1 | 497.0±46.9 | 0.16 | 0.08 |
| K D | Paddy | 0.6×103±0.4 | 43 | 42 | 15 | 7.3 | 0.39±31.7 | 277.2±15.7 | 0.14 | 0.11 |
| L U | Upland | 1.6×105±5.4 | 59 | 27 | 14 | 7.3 | 2.99±25.8 | 331.4±15.6 | 0.90 | 0.74 |
| M M | Upland | 2.6×103±1.9 | 40 | 30 | 30 | 7.3 | 2.36±79.3 | 604.4±26.8 | 0.39 | 0.25 |
| N D | Upland | N.D. | 45 | 37 | 18 | 7.5 | 4.50±35.4 | 2489.9±72.8 | 0.18 | 0.13 |
| O D | Upland | 1.6×104±5.2 | 52 | 22 | 16 | 7.4 | 0.61±102.3 | 220.8±31.2 | 0.27 | 0.17 |
| Correlation coefficientc | 0.598 (P<0.05) | −0.402 (P>0.05) | −0.567 (P<0.05) | −0.754 (P<0.01) d | −0.112 (P>0.05) | −0.259 (P>0.05) | 0.554 (P<0.05) | 0.541 (P>0.05) | ||
The lowest amount of yellow CFUs was detected in soil K with light contamination, while the upland soil L with the highest ratio of PAHs to total petroleum hydrocarbons (TPH) had the highest number of yellow CFUs. A significant correlation (P<0.05) was observed between the number of yellow CFUs and the ratio of PAHs to TPH (Table 1). It may be argued that the heavily PAH-polluted soils are too toxic for bacteria to survive, but this is certainly not the case for Sphingomonas. Sphingomonas are well-adapted to contaminated environments, especially those with high PAH to TPH ratios, indicating that members of the genus Sphingomonas are probably key members in natural PAH degradation. Indeed, many Sphingomonas strains have been isolated based on their ability to degrade a variety of PAHs, such as Sphingomonas sp. LB126 and S. paucimobilis EPA505.5,20
We also found that Sphingomonas seemed to preferentially associate with the sand fraction in the soil because the number of yellow CFUs had a significant (P<0.05) positive correlation with the proportion of the sand fraction. In contrast, a significant (P<0.05) negative correlation was found between the number of yellow CFUs and the clay fraction (Table 1). The association of the bacteria with polluted soil particles was shown previously. Uyttebroek et al.21 reported that Mycobacterium mainly accumulated in the PAH-enriched clay fraction, indicating that Mycobacterium might experience advantages connected to substrate source attachment. However, to the best of our knowledge no research has been performed on the number of culturable Sphingomonas in different soil fractions in contaminated soils. Bastiaens et al.20 compared two different procedures to isolate PAH-utilizing bacteria from PAH-contaminated samples and revealed that liquid enrichment mainly led to the isolation of Sphingomonas, while the membrane method exclusively led to the selection of Mycobacterium. Sphingomonas was postulated to prefer the sand habitat, in which the Sphingomonas were loosely associated with soil particles and thus obtained more oxygen to perform degradation.
The amounts of yellow CFUs in 4 paddy soils (B, C, D and H) with low pH values (6.4–6.8) were much higher than the amounts in the other 5 paddy soils (A, F, I, J and K) with relatively high pH values (7.3–8.1). Correlation analysis showed a significant decrease (P<0.01) in the amounts of yellow streptomycin-resistant CFUs with the increase in soil pH (the analysis was conducted with the 9 paddy soils that yielded yellow colonies) (Table 1). Thus, our data indicated that the culturable Sphingomonas preferred an acidic pH in petroleum-contaminated soils. Previous studies showed that Sphingomonas could survive in PAH-polluted soils with pH values ranging from 7.0 to 8.9,7,12,19,22 but the suitable soil pH for the removal of PAHs by Sphingomonas strains ranged between 6.0 and 7.0.6 This may explain to some extent why more culturable Sphingomonas were detected in soils with a relatively low pH value in this study. Nevertheless, the effect of soil pH on culturable Sphingomonas in petroleum-contaminated soils has not been previously reported. This finding raises further interest in the relationship between soil pH and the amounts of culturable Sphingomonas (especially the PAH-degrading isolates) in polluted soils.
Phylogenetic identification of isolatesSixty streptomycin-resistant yellow colonies were randomly selected from the 12 petroleum-contaminated soils and tested with Sphingomonas specific-PCR to exclude the non-Sphingomonas strains. A Sphingomonas species type strain Sphingomonas paucimobilis (1.3773, China General Microbiological Culture Collection Center) was used as the positive control. Simultaneously, 12 randomly chosen non-yellow pigmented colonies with different morphologies were tested together with the yellow colonies. No positive signals were displayed when Sphingomonas specific-PCR was performed on these non-yellow pigmented colonies (data not shown). A total of 48 out of the 60 yellow colonies produced amplicons of the expected size, indicating that the streptomycin medium together with the yellow pigment was only a semi-selective method for the identification of Sphingomonas species. The combination of this phenotypic detection method with Sphingomonas genus-specific PCR allowed genotypic confirmation of the yellow pigmented colonies, thereby facilitating the specific detection and isolation of culturable Sphingomonas from soil samples.
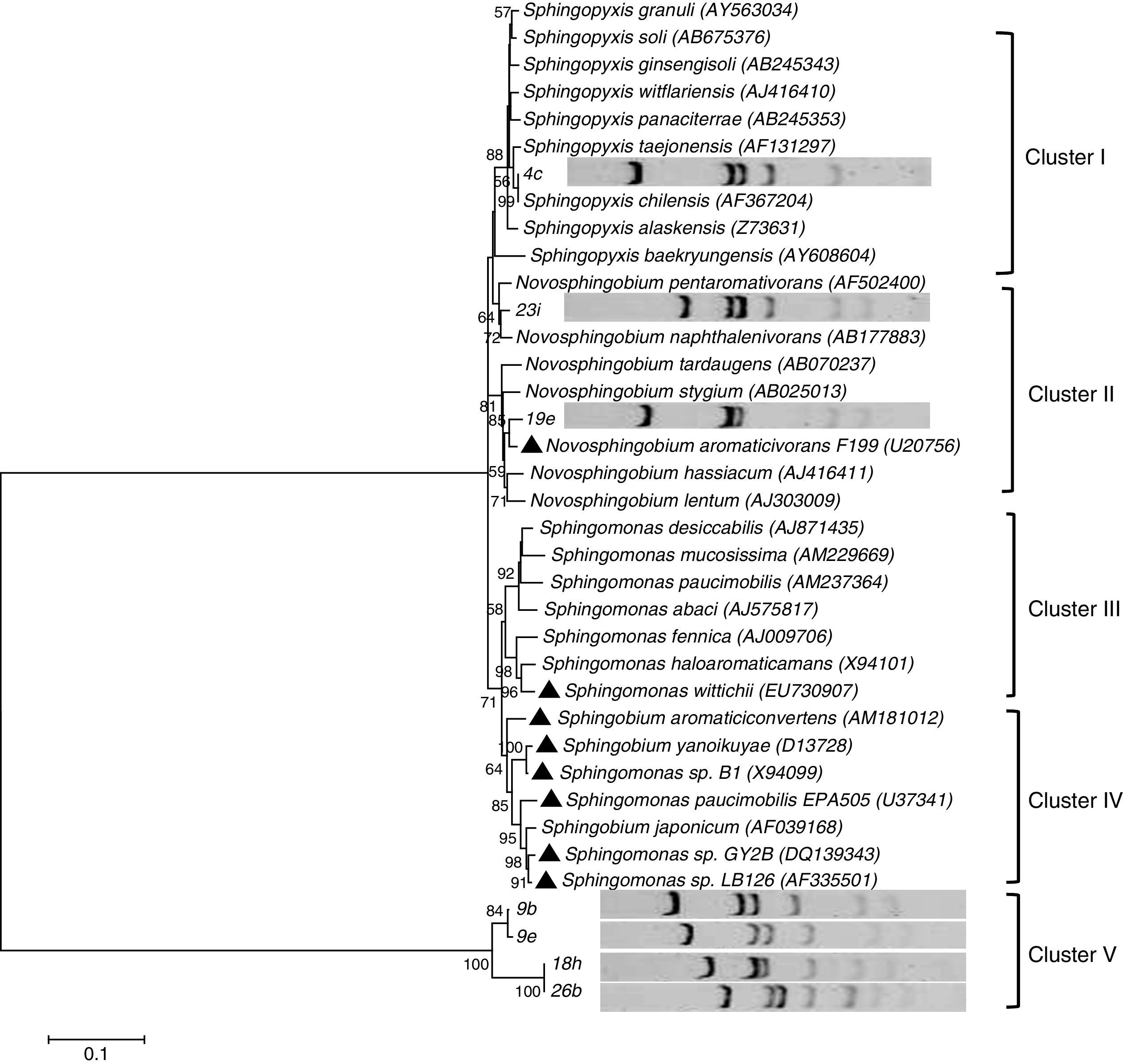
The 48 amplicons could be divided into 8 different phylotypes according to the RFLP analysis (Fig. 1). Sequence analysis showed that no sequence differences existed between the two representative strains from each phylotype; thus, only one strain was selected for further analysis. The results showed that 7 phylotypes matched significantly (99–100% similarities) with the Sphingomonas 16S rRNA gene sequences in GenBank. Of these 7 Sphingomonas phylotypes, two representative strains (9b and 9e) both shared similarities of 99% with Sphingomonas aromaticivorans (currently nominated as Novosphingobium aromaticivorans); however, RFLP analyses indicated that they were different strains (Fig. 1). One phylotype (2 out of the 48 total streptomycin-resistant yellow colonies) was not affiliated with Sphingomonas, but with Erythrobacter. It is not unexpected that Erythrobacter was detected together with Sphingomonas using the phenotypic and genotypic methods developed in this study because streptomycin resistance and yellow pigmentation have been reported previously for Erythrobacter strains (i.e., E. litoralis).12 Additionally, both Sphingomonas and Erythrobacter belong to the family Sphingomonadaceae, and the sequences of primer set SA429f/933r showed 100% similarity to the 16S rRNA gene sequences of Erythrobacter longus (data not shown). Thus, it can be inferred that the pheno- and phylo-methods developed in the present study that aimed to specifically isolate Sphingomonas members could inevitably select a minor fraction of other groups from Sphingomonadaceae.

Phylogenetic tree based on the alignment of 16S rRNA sequences of seven isolates and related sequences of the genus Sphingomonas was constructed using the neighbor-joining algorithm. The scale bar represents 0.1 substitutions per base position. The numbers at the nodes indicate the percentages of occurrence in 500 bootstrapped trees. Species harboring PAH-degrading isolates are indicated by “▴”. The accession numbers for selected species, which were suggested by Yabuuchi et al.4,23 for genus Sphingomonas and include Sphingomonas sensu stricto, Novosphingobium, Sphingobium, and Sphingopyxis, at this time are given in parentheses.
The phylogenetic tree (Fig. 1) constructed by aligning the isolated Sphingomonas sequences with sequences of typical species of genus Sphingomonas showed that only 3 representative strains (23i, 4c and 19e) clustered with known cultured Sphingomonas sequences. Strain 19e was closely related to Novosphingobium aromaticivorans F199, which could utilize fluorene and naphthalene as the sole carbon source24; this result suggested that strain 19e might have the capability for PAH degradation. The other 4 isolated strains (9b, 9e, 18h and 26b) were not placed in groups with known Sphingomonas but were grouped in a single cluster, indicating that they might represent new members of the genus Sphingomonas. Further analyses are needed to determine their physiological and biochemical characteristics.
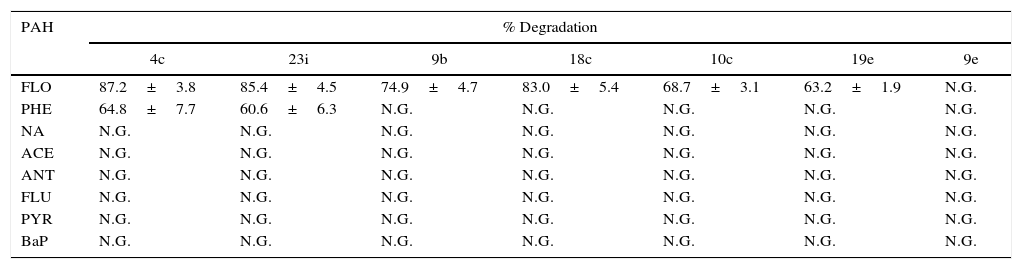
Utilization of PAHs as a sole source of carbon and energySeven Sphingomonas isolates representing 7 phylotypes were assessed for their ability to use 8 different individual PAHs as their sole carbon and energy source in liquid culture. Growth was considered positive when increases in turbidity were observed; positive growth responses were confirmed by analyzing the disappearance of the PAH with HPLC. Increases in turbidity and the disappearance of fluorene were observed in six of the seven tested strains (9b, 26b, 4c, 18h, 19e and 23i). Measurements of the degradation rates showed that Sphingomonas 4c could degrade 87.2% of the fluorene within 7 days at 30°C when the initial concentration of fluorene was 100mgL−1. Significant variation (P<0.01) in the fluorene degradation rate was observed among the 6 strains (Table 2). Additionally, strain 4c and strain 23i were capable of growing on phenanthrene (Table 2). Under the same incubation conditions, Sphingomonas 4c and Sphingomonas 23i could remove 64.8% and 60.6% of phenanthrene in the liquid culture, respectively. Overall, Sphingomonas strain 4c showed a capability to degrade fluorene and phenanthrene with a relatively high efficiency, indicating that it might serve as a promising functional strain for field studies of in situ bioremediation.
Degradation rate of eight PAHs by Sphingomonas strains.a
| PAH | % Degradation | ||||||
|---|---|---|---|---|---|---|---|
| 4c | 23i | 9b | 18c | 10c | 19e | 9e | |
| FLO | 87.2±3.8 | 85.4±4.5 | 74.9±4.7 | 83.0±5.4 | 68.7±3.1 | 63.2±1.9 | N.G. |
| PHE | 64.8±7.7 | 60.6±6.3 | N.G. | N.G. | N.G. | N.G. | N.G. |
| NA | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
| ACE | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
| ANT | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
| FLU | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
| PYR | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
| BaP | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. | N.G. |
NA – naphthalene; ACE – acenaphtene; FLO – fluorene; PHE – phenanthrene; ANT – anthracene; FLU – fluoranthene; PYR – pyrene; BaP – benzo[a]pyrene; N.G. – no growth.
The selected Sphingomonas isolates all failed to grow on the media supplemented with the other six PAH compounds as the sole carbon sources (Table 2). Although only 6 PAH-degrading Sphingomonas strains were obtained in the study, based on the fact that most of the bacteria were uncultured and the limitations of the isolation method it is reasonable to deduce that the diversity of PAH-degrading Sphingomonas strains in petroleum-contaminated soils is higher than observed in the present data.
The 6 PAH-degrading strains originated from 8 soil samples (A, C, D, H, I, K, L and O) with different contamination levels. Despite the fact that all of these soils contained a relatively high ratio of high molecular weight (HMW) PAHs to TPH (Table 1), none of the strains isolated in this study could utilize HMW-PAHs. The probable reason for this result may be explained by three facts. First, several studies have indicated that Sphingomonas are more adapted to the degradation of PAHs with relatively high bioavailability, such as phenanthrene.5,25,26 Second, the co-existence of bacteria/bacteria or bacteria/fungi known catabolic cooperation is ubiquitous in soil. Thus, we may speculate that Sphingomonas may be involved in HMW-PAH degradation by living on the metabolites of other bacteria or fungi rather than on the HMW-PAH itself. In the present study, degradation of HMW-PAHs was performed by pure strains but not by mixed cultures, which may provide a reduced chance of isolating HMW-PAH-degrading Sphingomonas strains. Last, our previous result suggested that the uncultivable Sphingomonas strains were the dominant ones in these contaminated soils11; hence, the uncultured Sphingomonas strains might play important roles in the degradation of HMW-PAHs.
Genetic analysis of several PAH-degrading pathways in Sphingomonas strains revealed the presence of a unique group of genes for aromatic compound degradation that were distantly related to those in other genera.24 Additionally, the catabolic genes from fluorene-utilizing Sphingomonas were unlike the other degradative genes involved in the degradation of other PAH components.27 The protocatechuic acid catabolic gene clusters found in the fluorene-degrading Sphingomonas sp. strain LB126 displayed a strong similarity with genes found in the lignin-degrading Sphingomonas paucimobilis strain SYK-6.24,27 However, little information is available concerning the identification of the dioxygenase genes involved in the first step of PAH degradation in Sphingomonas sp. LB126 or other Sphingomonas strains.28 Notably, the strains 9b and 9e were both identified as Sphingomonas aromaticivorans; however, only 9b (but not 9e) could degrade fluorene. It should be noted that not all Sphingomonas strains have the ability to degrade PAHs. This may reflect an ability of some Sphingomonas strains to more effectively recruit PAH-degrading genes as an adaptive response in PAH-contaminated soils.29 Moreover, this finding could indicate that different strains, even if they belong to the same species, might represent different functional groups. With the development of comparative genomics, similarities and differences between the genomes of strains 9b and 9e could be compared to reveal how the degrading genes were obtained or lost.
Conflict of interestThe authors declare that there is no conflicts of interest.
This work was supported by the National Natural Science Foundation of China No. 30800157 and the Strategic Priority Research Program of the Chinese Academy of Sciences XDB15010103.