





Amiloidosis es el término genérico utilizado para que hacer referencia al depósito en el tejido extracelular de fibrillas compuestas por fragmentos de bajo peso molecular de una gran variedad de proteínas. En condiciones normales, circulan de forma soluble como constituyentes del plasma, pero en ocasiones pueden sufrir un cambio conformacional de su estructura secundaria (plegándose con una morfología en lámina beta antiparalela) que las convierte en un producto insoluble que favorece su fragmentación en subunidades de bajo peso molecular. Estos fragmentos se depositan en el espacio extracelular de los tejidos como agregados nodulares ramificados constituyendo, tras la unión de una serie de cofactores que contribuyen a su estabilidad, el material amiloide1.
Aunque el material amiloide puede teñirse con diversas técnicas, la característica más típica es su tinción con rojo congo, tras lo cual adquiere birrefringencia verde manzana al exponerse a la luz polarizada. Esta característica se debe a la estructura química de los precursores proteicos que forman el amiloide, independientemente de su naturaleza, y es común a todos ellos2. En la mayor parte de las ocasiones, el subtipo de proteína amiloide únicamente se puede diferenciar con la utilización de técnicas inmunohistoquímicas.
La clasificación y la nomenclatura de los diferentes tipos de amiloidosis fue establecida por primera vez en el año 1998 por el Nomenclature Committee of the International Society of Amyloidosis (ISA). Desde entonces, los depósitos de material amiloide se clasifican utilizando la letra mayúscula A seguida por la letra de denominación de la proteína acumulada, sin espacio entre ambas. La actualización más reciente de esta nomenclatura fue realizada en el año 20143.
Amiloidosis cardíacaLa amiloidosis cardíaca (AC) es el depósito de material amiloide en el tejido extracelular cardíaco. Aparece más frecuentemente en el contexto de una afectación sistémica de la enfermedad, aunque también puede presentarse de forma localizada. La clínica cardiológica puede ser la primera manifestación, por lo que el cardiólogo presenta un papel muy relevante en su diagnóstico. Solo algunos subtipos de amiloidosis producen afectación cardíaca y cada uno de ellos lo hace con diferente severidad. Los 3 más frecuentes, en los que hay que centrar el diagnóstico diferencial, son: AL, ATTR (incluyendo sus 2 variantes, senil y hereditaria) y AA2,4.
Amiloidosis ALExiste un conjunto de enfermedades en las que se produce una proliferación inapropiada y exagerada de un único clon de células plasmáticas, por lo que reciben el nombre de discrasias de células plasmáticas o gammapatías monoclonales (GM). Estas células tumorales producen en grandes cantidades un único tipo de inmunoglobulina, denominado componente M. La amiloidosis AL está englobada dentro de las GM. En este caso, las células plasmáticas tumorales producen grandes cantidades del dominio variable de un único subtipo de cadena ligera (en el 75% de los casos de tipo lambda [λ]). Aunque poco prevalente, es la forma más frecuente de amiloidosis en los países desarrollados5. Como en otras GM, su incidencia aumenta a partir de la cuarta década de la vida, con una edad mediana de presentación de 64 años, y es más frecuente en varones.
La GM más frecuente es la GM de significado incierto (GMSI), representando el 60-70% del total y presentándose en el 5% de la población > 70 años. Por ello, la detección de un pico monoclonal durante el estudio diagnóstico de una amiloidosis no permite asumir que se trate de un tipo AL, especialmente en individuos de edad avanzada, puesto que podemos estar en frente de 2 entidades distintas de forma concomitante: una GMSI y una amiloidosis de etiología no AL6,7.
Los pacientes con amiloidosis AL presentan afectación cardíaca en un 50% de los casos, predominando en el cuadro clínico en el 20-30% y presentándose de forma aislada en un 4%. El órgano más frecuentemente afectado es el riñón, apareciendo algún grado de disfunción renal en 2/3 de los pacientes. Un 70% de los individuos presentan hepatomegalia y hasta un 2% desarrollará ictericia colostática, afección esta última que comporta un pésimo pronóstico. La neuropatía periférica, presente en un 20% de los pacientes, es una polineuropatía sensitivo-motora lentamente progresiva similar a la diabética. Otras afectaciones neurológicas características son el síndrome de túnel carpiano (10-20%) y la neuropatía autonómica (15%), responsable esta última de complicaciones severas y limitantes, como la disfunción eréctil o la hipotensión ortostática. La presencia de macroglosia o de púrpura periorbitaria son muy características y su combinación muy indicativa de amiloidosis AL, pero se presentan únicamente en un 10-15% de los casos.
Amiloidosis ATTRLa proteína precursora en esta ocasión es la transtirretina (TTR), también denominada prealbúmina (debido a que presenta un recorrido más rápido en comparación con la albúmina en los geles de electroforesis). Es una molécula con morfología tetraédrica y de síntesis principalmente hepática (un 98% del total, produciéndose el 2% restante en los plexos coroideos) que circula por la sangre y el líquido cefalorraquídeo actuando como transportador de la hormona tiroxina (T4) y la proteína de unión al retinol. Es precisamente de su función de donde deriva su nombre: transports thyroxine and retinol.
Tanto la proteína nativa como una forma mutada pueden actuar como precursoras en la formación del material amiloide. En el primer caso, nos encontramos ante una amiloidosis ATTR senil y en el segundo, ante una amiloidosis ATTR familiar. La primera suele presentarse en individuos > 60-70 años y en la totalidad de los casos existe afectación cardíaca; en la segunda, tanto la edad de presentación como la probabilidad de compromiso cardíaco varían en función del tipo de mutación que se trate.
Los depósitos amiloides sin significación clínica a nivel cardíaco son muy frecuentes en los ancianos. Pero en ocasiones estos depósitos son masivos y generan un compromiso de la función cardíaca con repercusión clínica. En la amiloidosis ATTR senil, la afectación cardíaca es universal y más frecuentemente ocurre de forma aislada. En un escaso porcentaje de pacientes pueden verse afectados también otros órganos, siendo los más frecuentes el pulmón, el riñón y los nervios periféricos.
La amiloidosis ATTR familiar es una enfermedad genética, con herencia autosómica dominante y elevada penetrancia. El gen que codifica esta proteína se encuentra en el cromosoma 18 y existen más de 120 mutaciones descritas. El órgano afectado de forma predominante va a depender del tipo de mutación, existiendo 14 formas que se caracterizan por el compromiso cardíaco.
Amiloidosis AAEste tipo de amiloidosis se presenta en el contexto de enfermedades inflamatorias o infecciosas crónicas y neoplasias, y es la forma más frecuente en países en vías de desarrollo. La proteína precursora es el amiloide A sérico, una apolipoproteína de síntesis hepática que actúa como reactante de fase aguda. El órgano más precoz y frecuentemente afectado es el riñón (80% de los pacientes), apareciendo habitualmente también hepatomegalia y síntomas derivados del depósito amiloide en el tubo digestivo. La afectación cardíaca es poco frecuente (5%) y generalmente asintomática.
Manifestaciones clínicas derivadas de la afectación cardíacaLa infiltración del tejido extracelular cardíaco por el material amiloide provoca un incremento del grosor del órgano, lo que lleva a un aumento de su rigidez y una alteración progresiva de la función diastólica. Esto implica la aparición de síntomas y signos de insuficiencia cardíaca (IC), tanto izquierda como derecha, dado el compromiso biventricular. En fases avanzadas, la infiltración amiloide facilita la necrosis miocitaria y su sustitución por tejido fibrótico, desembocando en un proceso de remodelado cardíaco con dilatación de las cavidades y alteración de la función sistólica.
El material amiloide infiltra aurículas y ventrículos por igual, lo que puede generar una disfunción electromecánica auricular que favorece el estasis sanguíneo y aumenta el riesgo de formación de trombos, incluso en ritmo sinusal8. Por ello, los pacientes con AC tienen alto riesgo de fenómenos tromboembólicos, especialmente aquellos con amiloidosis AL o fibrilación auricular9.
Los depósitos amiloides también infiltran las arterias coronarias, habitualmente afectando a la microvasculatura y respetando los vasos epicárdicos. Esto genera isquemia y necrosis miocitaria que pueden expresarse en forma de angina y también contribuyen al remodelado cardíaco citado previamente10.
Los episodios presincopales y sincopales son frecuentes. Existen varios mecanismos que participan en su aparición, como la neuropatía autonómica o la incapacidad del corazón infiltrado para mantener un gasto cardíaco adecuado. Este último es el responsable en la mayor parte de los casos de la aparición de episodios sincopales con el esfuerzo, marcador de mal pronóstico y elevada mortalidad a corto plazo11. En ocasiones, los eventos arrítmicos subyacen como responsables de este tipo de eventos, tratándose generalmente de bradiarritmias y siendo las arritmias ventriculares excepcionales protagonistas12.
Evaluación inicial del paciente con amiloidosis cardíacaExploración físicaAunque puede ofrecernos en ocasiones una serie de hallazgos que indiquen el diagnóstico, la mayor parte de las veces va a resultar inespecífica y compatible con muchas otras causas de IC. El dato semiológico más específico es la coexistencia de macroglosia y una púrpura periorbitaria, resultando su presencia en un paciente con IC prácticamente patognomónica de amiloidosis, concretamente del tipo AL.
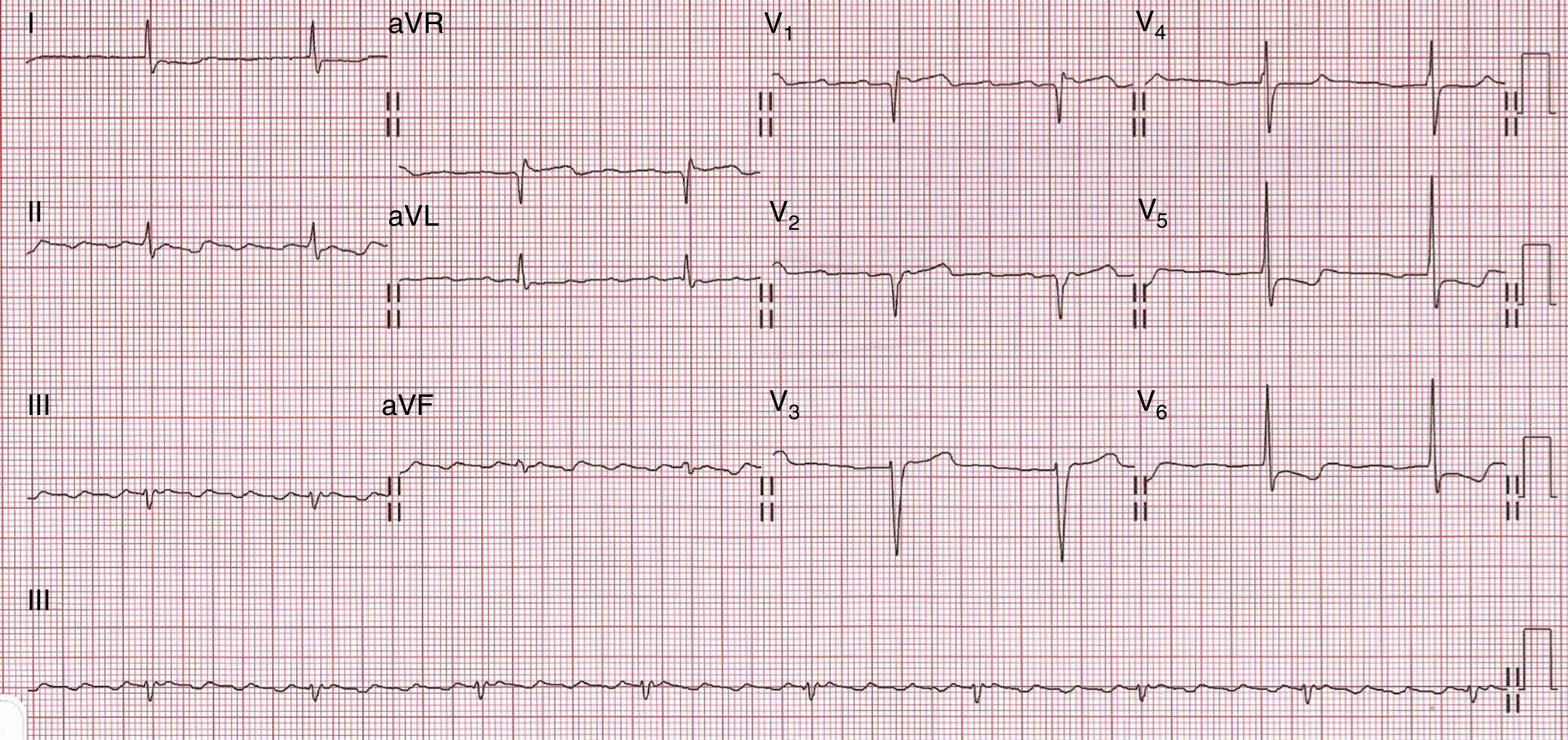
Electrocardiograma de 12 derivacionesLa alteración electrocardiográfica asociada clásicamente con una mayor rentabilidad diagnóstica en la AC es la presencia de bajos voltajes en las derivaciones de los miembros (definido como un QRS ≤ 0,5mV) (fig. 1). Sin embargo, este hallazgo se encuentra únicamente en un 50% de los pacientes con amiloidosis AL, en un 40% de los que presentan amiloidosis ATTR senil y en un 25% de aquellos con amiloidosis ATTR familiar. Otros parámetros utilizados para definir la presencia de bajos voltajes en el ECG son la existencia de un QRS ≤ 1mV en las derivaciones precordiales o un índice de Sokolow-Lyon<1,5mV. Un estudio publicado recientemente13 ha encontrado que este último parámetro resulta el más rentable para el diagnóstico de la AC, presentando una prevalencia de aparición del 60% y sin existir diferencias entre los diferentes subtipos de amiloidosis.
.")
Otras alteraciones electrocardiográficas que nos podemos encontrar son un patrón de seudoinfarto (20-50%, siendo el más frecuente anteroseptal) (fig. 2), un intervalo PR prolongado (20-30%) e incluso criterios de crecimiento ventricular izquierdo (10-15%).
Pruebas diagnósticas en la amiloidosis cardíacaEcocardiograma transtorácico
El ecocardiograma transtorácico (ETT) es la prueba de elección para iniciar el proceso diagnóstico, debiendo tener en cuenta que los diferentes subtipos de la enfermedad presentan hallazgos indistinguibles.
La alteración más precoz es un incremento del grosor de la pared biventricular, con la consiguiente alteración de la función diastólica. El patrón generalmente es concéntrico y simétrico, siendo poco común el acúmulo asimétrico del material amiloide a nivel del septo interventricular (5% de los casos)14. Con la progresión de la enfermedad también aumenta el grado de disfunción diastólica, llegando a presentarse incluso en estadios avanzados un compromiso de la función sistólica.
La presencia de derrame pericárdico es muy frecuente (40-60%), siendo generalmente de grado ligero y sin repercusión clínica.
La infiltración miocárdica por el material amiloide provoca un aumento parcheado de su ecogenicidad, dándole un aspecto ecocardiográfico granulado (sparkling) que ha sido propuesto como un dato muy indicativo de esta enfermedad. Sin embargo, este patrón se encuentra únicamente en un 26-36% de los pacientes, lo que lo convierte en un hallazgo poco útil para el diagnóstico de la AC15.
Las técnicas de strain también resultan de utilidad en la evaluación diagnóstica de la AC. De forma característica, se produce una reducción de la contractilidad longitudinal que es mucho más marcada que la presente en otras cardiopatías que cursan con engrosamiento ventricular, como la miocardiopatía hipertrófica. También se produce un patrón típico de alteración de la contractilidad radial, caracterizado por una afectación marcada a nivel basal con normalidad de los parámetros de deformidad apicales16.
En resumen, los hallazgos ecocardiográficos en muchos casos van a resultar inespecíficos y comunes a otras cardiopatías. La combinación de estos con la información aportada por el electrocardiograma goza de una mayor rentabilidad diagnóstica, presentando una sensibilidad de entre el 72 y el 79%, y una especificidad del 91-100%.
Resonancia magnética nuclearAdemás de una mejor caracterización de la morfología cardíaca, la resonancia magnética nuclear (RM) tiene la ventaja añadida respecto al ETT de poder obtener imágenes de realce tardío con gadolinio (RTG), técnica que permite indicar fuertemente el diagnóstico de AC en gran parte de los casos (alcanzando una sensibilidad del 86-88% y una especificidad del 86-90%). Se han descrito varios patrones de RTG indicativos de esta enfermedad, pero es el realce subendocárdico global en anillo el que se presenta con más frecuencia y el que clásicamente se ha asociado a la AC17. La mayor cantidad de información obtenida con la RM permite una detección más precoz de la enfermedad y facilita el diagnóstico diferencial con otras entidades que cursan con un aumento del grosor miocárdico.
En los últimos años, se ha desarrollado una técnica para el estudio de la AC que presenta la ventaja de no precisar administración de gadolinio, conocida como T1-mapping. Con ella es posible una detección más precoz de la enfermedad en comparación con el estudio del patrón de RTG, permite la cuantificación del depósito amiloide y sirve de ayuda en el diagnóstico diferencial entre los subtipos de amiloidosis AL y ATTR18.
Imagen nuclearEsta técnica se fundamenta en la utilización de radiofármacos, compuestos constituidos por 2 componentes: una molécula ávida por un tejido concreto y un radiofármaco, cuya actividad radiactiva es posible detectar.
Al inicio de la década pasada, comenzó a estudiarse la utilidad potencial del 99mTc asociado a otra molécula, el ácido 3,3-difosfono-1,2-propanodicarboxílico (99mTc-DPD). Esta molécula se deposita en aquellos tejidos con un alto contenido en calcio, pero también se caracteriza por presentar una gran avidez por los depósitos amiloides del subtipo ATTR. El primer estudio con este fármaco demostró una captación cardíaca del radiofármaco en la totalidad de los pacientes con amiloidosis ATTR demostrada y en ninguno de los que presentaban el subtipo AL o pertenecían al grupo de controles sanos19. Sin embargo, esta diferenciación no siempre es tan marcada, habiéndose comprobado que hasta un tercio de los pacientes con amiloidosis AL pueden presentar captación ligera del radiofármaco a nivel cardíaco20. Debido a esto, se ha propuesto una clasificación en 4 grupos en función de la intensidad de la captación de la molécula:
- –
Grado 0: ausencia de captación cardíaca, captación ósea normal.
- –
Grado 1: captación cardíaca ligera, menor que la captación ósea.
- –
Grado 2: captación cardíaca moderada, mayor que la captación ósea que se encuentra atenuada.
- –
Grado 3: captación cardíaca intensa, captación ósea ligera o ausente.
Así, se consideran los grados 2 y 3 indicativos de amiloidosis ATTR, pudiendo presentarse los grados 0 y 1 en el resto de subtipos de AC e individuos sanos.
Biopsia tisularLa demostración histológica de la presencia del depósito amiloide es una condición sine qua non para el diagnóstico definitivo de la enfermedad. La muestra tisular puede obtenerse de órganos sin expresión clínica (p. ej., grasa subcutánea abdominal, mucosa rectal o glándulas salivales menores), siendo esta opción un procedimiento seguro pero con baja sensibilidad en los casos de amiloidosis localizada, por lo que debe reservarse para pacientes con afectación multiorgánica. Alternativamente, puede plantearse la toma de la muestra de un órgano afectado clínicamente (p. ej., corazón o riñón). En este caso, el riesgo de complicaciones es mayor, pero aumenta la capacidad diagnóstica. Esta opción debería plantearse en los casos sin afectación sistémica o si la sospecha diagnóstica persiste a pesar de un resultado negativo de una biopsia obtenida de un tejido sin expresión clínica.
El examen microscópico de la muestra del tejido afectado evidencia la presencia de depósitos de material hialino amorfo localizados predominantemente en el espacio extracelular. Se tiñe con diferentes tinciones (tioflavinaT, azul alcián), pero es la captación de rojo congo y la adquisición de una birrefringencia verde manzana al ser sometido al microscopio de luz polarizada lo que resulta más definitorio.
Diagnóstico del subtipo de amiloidosis cardíacaUna vez confirmado el diagnóstico, es fundamental establecer el subtipo, puesto que el tratamiento etiológico difiere. Para ello, a lo largo del proceso diagnóstico deberemos solicitar una serie de pruebas complementarias con esta intención.
Determinación del componente monoclonalDado que el subtipo de amiloidosis AL es el más frecuente en nuestro medio, la batería de pruebas complementarias que se realizan a un paciente con sospecha de la enfermedad debe incluir la determinación del componente monoclonal. Para ello, ha de solicitarse una electroforesis de sangre y orina (permite la detección y la cuantificación del componente monoclonal, siendo positivo en un 50% de los casos), un estudio de inmunofijación de sangre y orina (permite la detección y tipificación del componente monoclonal, siendo positivo en un 90% de los casos) y un estudio de cuantificación por nefelometría (Freelite®) de cadenas ligeras en sangre (positivo en un 99% de los casos)21. Aunque la positividad de esta determinación en el contexto de un paciente con otros datos indicativos de amiloidosis apoya el diagnóstico de la enfermedad, y en concreto del subtipo AL, por sí sola no permite la confirmación definitiva. Este hecho se ilustra con los resultados de un estudio que incluyó a 350 pacientes con confirmación histológica de amiloidosis, ausencia de historia familiar de la enfermedad y la presencia de componente monoclonal. Hasta un 10% de ellos presentaban una mutación del gen de la proteína TTR6. Por tanto, es relativamente frecuente encontrar un pico monoclonal en pacientes, generalmente de edad avanzada, con amiloidosis. Ello no puede llevar por sí solo a asumir el diagnóstico de un subtipo AL, puesto que en un porcentaje no desdeñable de casos podemos estar delante de una GMSI que se presenta de forma concomitante con una amiloidosis no AL7.
Imagen nuclearLos estudios nucleares con 99mTc-DPD que resultan positivos son altamente específicos para un subtipo de amiloidosis, la ATTR, por lo que esta prueba desempeña un papel fundamental en el diagnóstico diferencial.
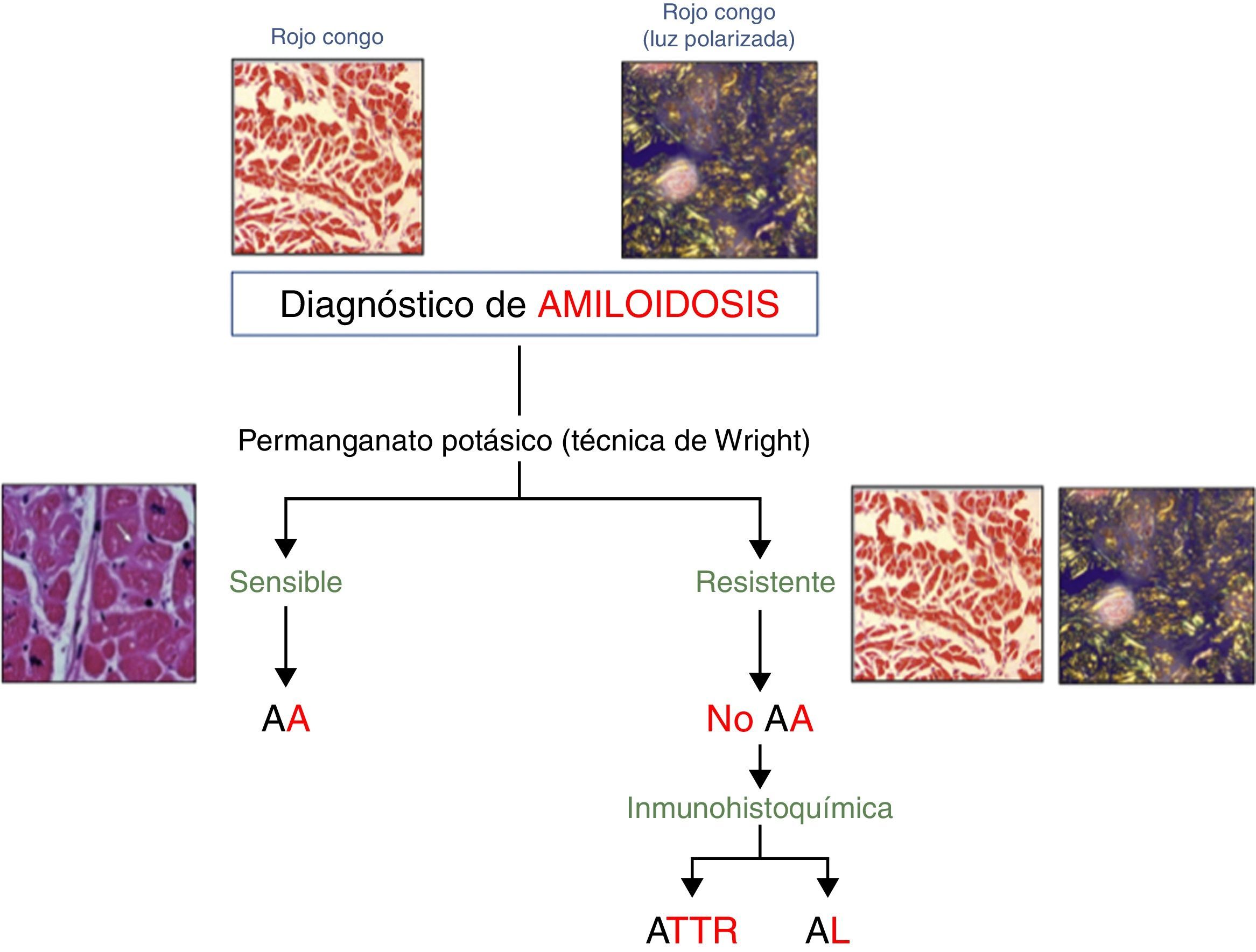
Tinciones histológicasLa confirmación definitiva del subtipo de amiloide únicamente vamos a obtenerla con el estudio anatomopatológico. Tras confirmar la presencia de material amiloide con la tinción de rojo congo, se somete a la muestra a una solución de permanganato potásico (técnica de Wright). Si tras ello el tejido ha dejado de teñirse, se tratará de un depósito amiloide de tipo AA; por el contrario, si persiste la tinción de rojo congo, nos encontraremos ante un depósito amiloide de tipo no AA. Para diferenciar entonces el subtipo de amiloidosis, hay que recurrir a tinciones específicas de inmunohistoquímica, que van a establecer el diagnóstico en un amplio porcentaje de casos (fig. 3). Sin embargo, la sensibilidad y la especificidad de estas tinciones no alcanzan el 100%, por lo que en casos equívocos debe de utilizarse una técnica de microdisección con láser seguida de una espectrometría de masa, técnica que alcanza una mayor rentabilidad diagnóstica22.
Algoritmo diagnóstico
Para finalizar, exponemos a continuación el algoritmo que es utilizado durante el proceso diagnóstico de la AC en nuestro centro, elaborado con la intención de protocolizar los pasos a seguir ante la sospecha clínica de esta entidad (fig. 4).
Responsabilidades éticasProtección de personas y animales
Los autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que en este artículo no aparecen datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

