




Un porcentaje no despreciable de pacientes con cardiopatías presentan una enfermedad directamente condicionada por una causa genética. En los últimos 10años se ha producido una explosión del conocimiento de las bases moleculares de estas enfermedades gracias al desarrollo de técnicas de secuenciación del genoma cada vez más rápidas y eficientes. Este gran avance ha hecho realidad que podamos disponer para uso clínico del estudio genético y así conocer la mutación que origina la enfermedad. Este hecho supone un cambio radical en el abordaje de estos pacientes y de sus familias.
En base a esto, todo cardiólogo debería conocer al menos una serie de conceptos básicos sobre genética para poder aproximarse de forma adecuada a estos pacientes y a sus familias.
¿Qué conceptos de genética debe conocer un cardiólogo?El ácido desoxirribonucleico (ADN) es un polímero de nucleótidos que constituye el material genético de nuestras células. Este ADN se organiza en los cromosomas. Los seres humanos disponemos de 23pares de cromosomas.
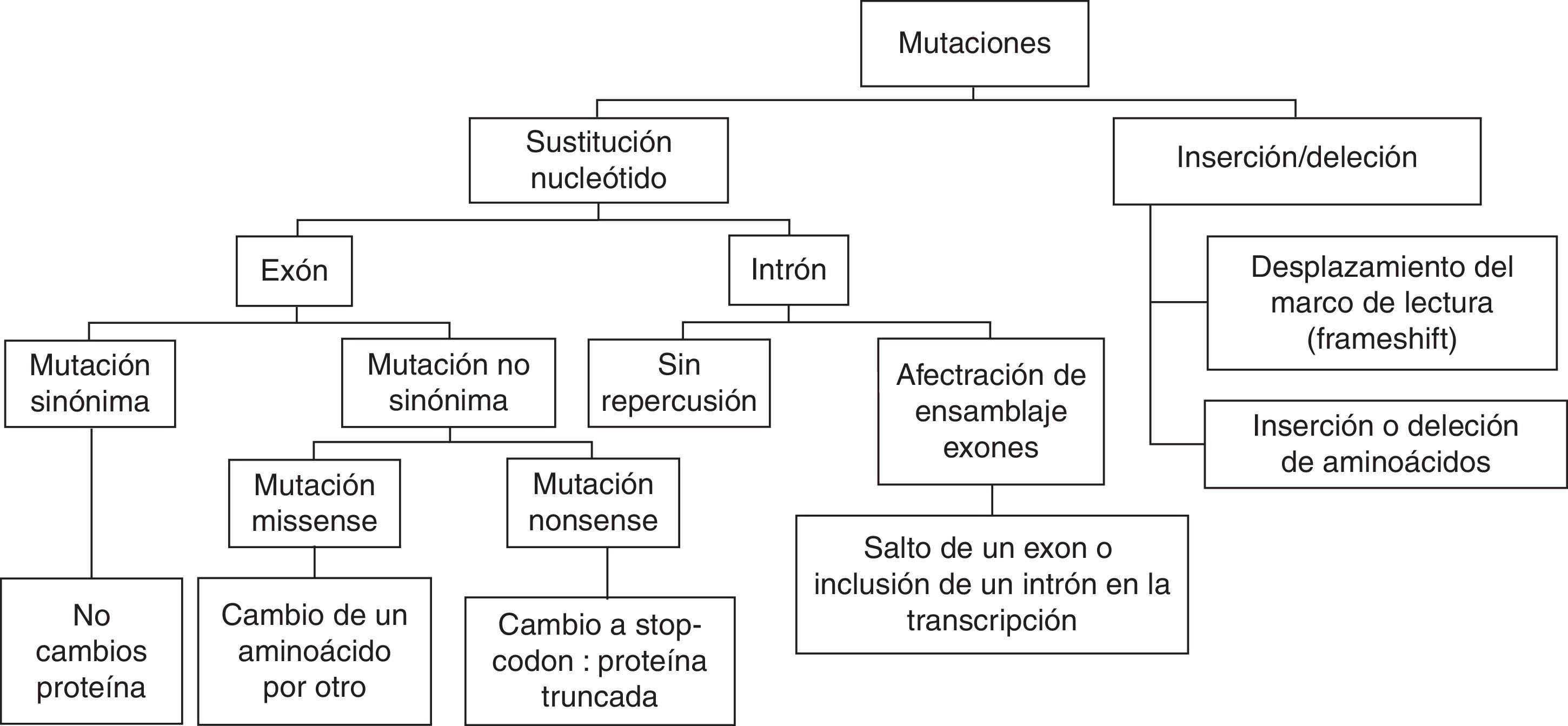
Llamamos gen a cada porción de ADN que codifica para una proteína. La secuencia de nucleótidos de un gen se divide en exones e intrones. Los exones son las partes del gen que codifican para proteínas. Entre los exones existen intrones: secuencias de nucleótidos más o menos largas de las que se prescinde para la síntesis proteica. Esta exclusión de los intrones se realiza en el proceso de transcripción del ADN al ARN mensajero. En los exones, cada 3 nucleótidos (triplete o codón) determinan un aminoácido en la síntesis proteica.
Las células somáticas (no germinales) de los seres humanos son diploides, es decir disponen de 2 copias para cada gen, una procedente de la madre y otra del padre. A cada copia se le denomina alelo. La mayor parte de la secuencia de nuestro ADN, alrededor del 99%, es igual al ADN del resto de personas. Sin embargo, pueden existir pequeñas diferencias en la secuencia de nucleótidos entre unos individuos y otros. Cuando estas variaciones son relativamente frecuentes en la población general (>0,5%), y habitualmente no influyen en la estructura o función de las proteínas resultantes, se suelen denominar polimorfismos. Hay excepciones de variantes de secuencia que a pesar de tener una frecuencia muy baja en la población, no producen ningún efecto patogénico, denominándose comúnmente variantes raras (rare variants). En general los polimorfismos más frecuentes son las sustituciones nucleotídicas (Single Nucleotide Polymorphisms [SNP]) o las variaciones en el número de copia de ciertas repeticiones de polinucleótidos (Short Tandem Repeats [STR]). La presencia de estos polimorfismos a nivel génico conduce a la existencia de diversos alelos para cada gen, que difieren únicamente en la combinación de variantes que presentan. En cambio, si las variantes de secuencia dan lugar a una alteración a nivel proteico que a su vez conduce a una modificación en la funcionalidad de la proteína, confiriéndole un efecto patogénico, se utiliza el término mutación. Las mutaciones que se suelen encontrar en patología cardiovascular se reflejan en la figura 1. Las más frecuentes son las sustituciones de un nucleótido por otro. En términos de diagnóstico genético de enfermedades hereditarias, una de las grandes dificultades actuales es la distinción entre una mutación patogénica en el contexto de una patología determinada, y una variante rara sin significado patogénico.

Por genotipo entendemos la secuencia del ADN de una persona, incluyendo las variantes genéticas que porta, entre las que podría estar una mutación patogénica. Fenotipo es la expresión clínica del genotipo, que puede estar influida por el medio. El concepto de penetrancia, aplicado a las enfermedades autosómicas dominantes (ver más abajo), es la probabilidad de que una persona que porta una mutación patogénica exprese el fenotipo de enfermedad. Expresividad es el grado diverso de expresión que pueden manifestar en el fenotipo distintos individuos con la misma mutación.
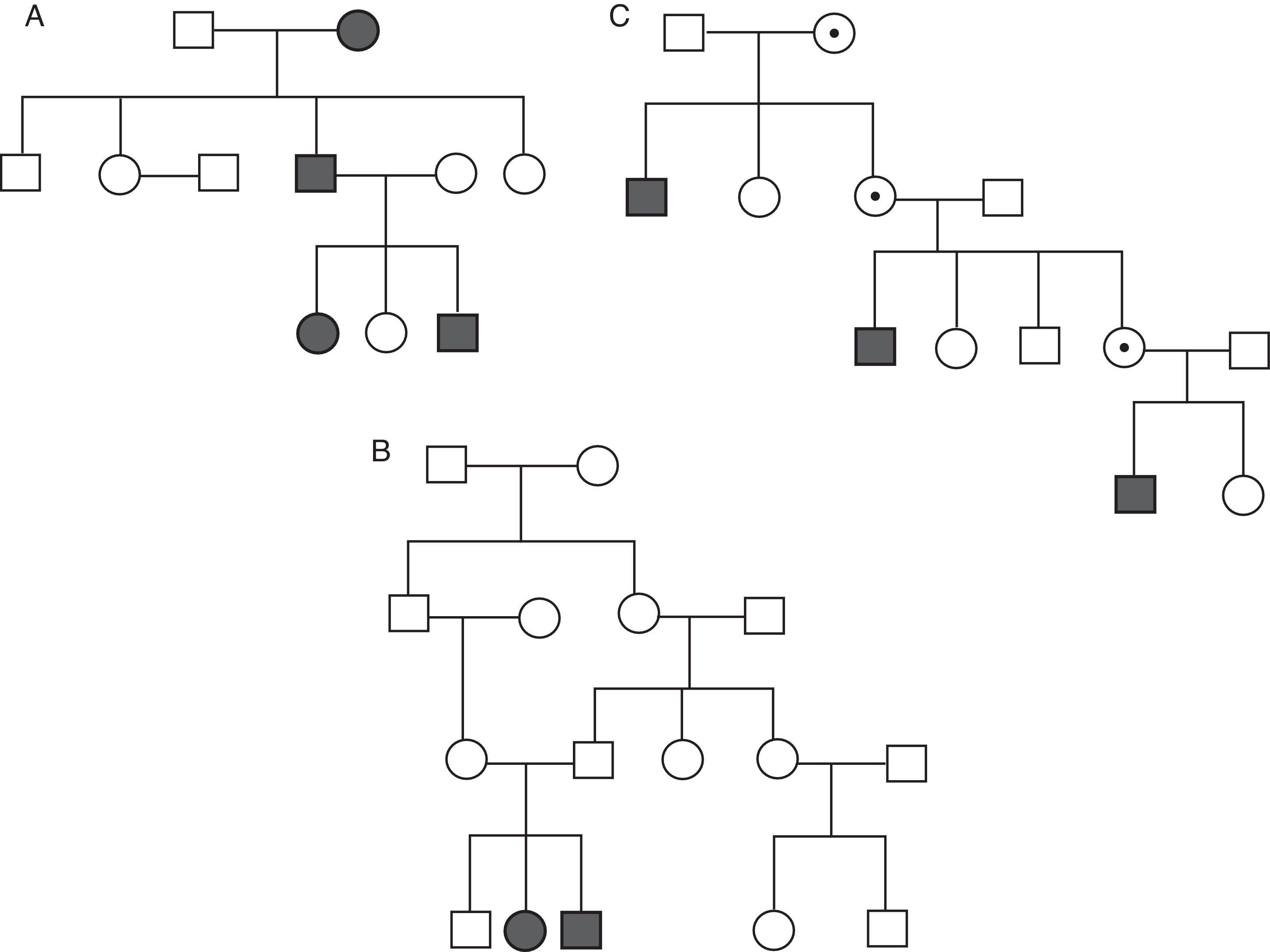
Las enfermedades monogénicas pueden tener distintos tipos de patrones de transmisión (fig. 2): autosómico dominante, autosómico recesivo o ligado al cromosomaX. En el caso de herencia autosómica dominante es suficiente un alelo mutado para que se exprese el fenotipo de la enfermedad. En las enfermedades autosómicas recesivas ambos alelos han de estar mutados para que la enfermedad se exprese, aunque en algunos casos los portadores heterocigotos pueden tener formas leves de la enfermedad y estar oligosintomáticos. La herencia ligada al cromosomaX puede ser también dominante o recesiva. En el primer caso tanto varones como mujeres padecen la enfermedad, pero la expresión en varones es más severa. En el segundo caso solo los varones expresan la enfermedad, mientras que las mujeres son portadoras o, en el peor de los casos, oligosintomáticas. La mayoría de las enfermedades cardiovasculares hereditarias responden a un patrón de herencia autosómico dominante. Sin embargo, el patrón de herencia puede ser la clave para la sospecha diagnóstica en ciertas situaciones como la enfermedad de Fabry, de herencia ligada al cromosomaX, cuyo fenotipo puede confundirse con una miocardiopatía hipertrófica pero que tiene tratamiento específico con reemplazo enzimático. Para poder apreciar el tipo de herencia es obligada la confección de un árbol genealógico de al menos 3 generaciones en el estudio del caso índice y los familiares.
¿Cómo se hace y cómo se interpreta un estudio genético?
La gran revolución en el diagnóstico molecular de las cardiopatías familiares ha sido el desarrollo de plataformas que permiten la secuenciación del ADN de una manera muy rápida, exhaustiva y coste-eficiente. Esta nueva tecnología, conocida como Next Generation Sequencing (NGS), ha sustituido la aproximación del estudio genético desde un concepto de linkage genético y secuenciación de un limitado número de genes candidatos mediante secuenciación first-generation a un concepto de secuenciación de paneles de genes según patología que incluyen un amplio número de genes1. El uso de paneles de secuenciación multigenes se ha demostrado necesario en las cardiopatías heredables, debido a la gran heterogeneidad de genes y de mutaciones dentro de un mismo gen que son capaces de provocar el mismo fenotipo.
Con esta nueva tecnología, el talón de Aquiles del análisis genético es la interpretación y la traslación clínica de las variantes halladas en el estudio. Ante el hallazgo de una mutación no frecuente en la población general, el punto clave es distinguir si se trata de una mutación patogénica o de una variante rara «benigna». Debe pensarse que una mutación es patogénica cuando se segrega con el fenotipo en otras familias donde haya sido encontrada la misma mutación. Sin embargo, muchas mutaciones aparecen por primera vez en una familia («privadas»). Las mutaciones que producen proteínas truncadas tienen más probabilidad de ser patogénicas, como es el caso de las mutaciones nonsense, que consisten en cambios puntuales que conducen a la sustitución de un aminoácido por un codón de parada, o de las deleciones o duplicaciones que conducen a una alteración de la secuencia proteica y a terminaciones prematuras de la proteína resultante. Sin embargo, la mayoría de las variantes encontradas son mutaciones tipo missense, donde se produce solo un cambio de un aminoácido por otro en la proteína (fig. 1). El estudio in vitro de la repercusión de la mutación en la función proteica puede ser útil, aunque tiene sus limitaciones y no es metodológicamente factible realizarlo para cada nueva variante diagnosticada. En análisis in silico mediante programas bioinformáticos para analizar la conservación del aminoácido mutado a lo largo de las especies, o para determinar si la mutación se ubica en un dominio funcional de la proteína, puede añadir información. Así, por ejemplo, en el síndrome del QT largo el estudio funcional ha permitido establecer la probabilidad patogénica de una mutación en función del dominio donde se localice la variante2. Finalmente, la disponibilidad de bases de datos con el genoma de poblaciones numerosas puede proveer de información sobre la frecuencia de una mutación en la población de la misma etnia del sujeto en estudio, de manera que cuanto menos frecuente sea la mutación más probable es que esta sea patogénica. Pese a todo esto, en ocasiones no tendremos la suficiente información y certeza sobre si una mutación puede ser causante de la enfermedad en el paciente estudiado, en cuyo caso denominaremos a la mutación como una variante de significado incierto. La más apropiada aproximación al resultado de un test genético es interpretarlo de una forma probabilística, en lugar de una forma determinista3.
En los últimos años se han publicado recomendaciones sobre las indicaciones de estudio genético en las distintas miocardiopatías y canalopatías4.
En nuestro país disponemos de varios laboratorios que ofrecen la realización de estudio genético para cardiopatías familiares. Además de cumplir unos estándares de calidad en el análisis y la secuenciación del ADN, es muy deseable para el cardiólogo que el informe final del laboratorio no sea únicamente un informe molecular con una simple relación de variantes o mutaciones, sino que la mutación se coteje con las mutaciones ya publicadas y con bases de datos propias o ajenas, de manera que se ofrezca una interpretación clínica de la anomalía molecular.
No hay que olvidar que una buena indicación e interpretación de un estudio genético comienza por un adecuado estudio clínico del caso índice y de los familiares de primer grado, así como por la elaboración de un completo árbol genealógico que recoja los antecedentes familiares con una atención especial a la ocurrencia de muerte súbita. El análisis genético se realizará en primer lugar al familiar con mayor expresividad fenotípica de la enfermedad, ya que es aquí donde existe la mayor probabilidad de identificar una mutación patogénica. En el caso de hallar una mutación, esta será específicamente buscada en los familiares de primer grado. El diagnóstico de un fenotipo y/o genotipo positivo en un familiar llevará consigo la extensión del estudio a los familiares de primer grado de este (estudio en cascada).
¿Qué es el consejo genético y cómo se lleva a cabo?El consejo genético es el conjunto de actuaciones que tiene como objetivo ayudar al paciente y a su familia a comprender y adaptarse a las implicaciones médicas, psicológicas y familiares que conlleva el diagnóstico de una enfermedad genética5. El hecho de que el denominador común de muchas de las cardiopatías familiares sea un hecho tan devastador como la posibilidad de una muerte súbita subraya la importancia de una información y soporte emocional adecuados.
Las actuaciones que han de proveerse en el consejo genético se resumen en la tabla 1. El consejo genético no supone la realización de un análisis genético ni se aplica únicamente cuando este está disponible, sino que forma parte del manejo de estos pacientes y sus familias desde el momento en que se realiza el diagnóstico clínico en el caso índice. El consejo genético pretende a través de una adecuada información facilitar el bienestar psicológico y reducir la ansiedad que para sí y sus familiares presentes y potencialmente futuros presentan los pacientes.
Puntos clave del consejo genético
| Información | Apoyo psicológico | Planificación familiar |
| Posibilidad de otros familiares afectos | Apoyo ante el dolor por la muerte de familiares | Probabilidad de que la descendencia herede la enfermedad, implicacionesRiesgos en la gestación para la madre con enfermedadGestaciones ya concebidas: posibilidad de diagnóstico fetalConsejo genético preconcepcional: posibilidad de diagnóstico embrionario preimplantacional (selección embriones sin mutación) |
| Modo y riesgo de heredar la enfermedad | ||
| Estudio clínico a familiares | ||
| Fenotipo(+):• Cambio estilo vida (deportes, evitar fármacos…)• ¿Necesidad de terapia?• ¿Riesgo de muerte súbita? → ¿fármacos, DAI? | Preocupaciones y miedos ante el diagnóstico y ante el riesgo de MS | |
| Fenotipo(−):• Posibilidad de desarrollo futuro de la enfermedad → seguimiento clínico periódico | Preocupación por los familiares → sentimiento de culpa, temor, enfado, etc. | |
| Estudio genético: informar de indicación, ventajas y limitaciones | Cambio de percepción en el estado de salud | |
| Fenotipo(+)/Genotipo(+):• Confirmación diagnóstica vs fenocopia (MH) → ¿tratamiento específico?• ¿Mayor información pronóstica? (sd. del QT largo) | Impacto psicológico del cambio de estilo de vida (deporte de competición en jóvenes) | |
| Fenotipo(−)/Genotipo(+):• Posibilidad de desarrollo futuro de la enfermedad → seguimiento clínico periódico | Recursos destinados a grupos de pacientes | |
| Fenotipo(−)/Genotipo(−):• Alta |
DAI: desfibrilador automático implantable; MH: miocardiopatía hipertrófica; MS: muerte súbita.
A la hora de indicar el estudio genético es preciso explicar a la familia su utilidad, sus ventajas y sus limitaciones, evitando que la decisión final sobre su realización se base en expectativas irreales. El principal impacto clínico que la detección de la mutación causante va a tener en la mayoría de las patologías cardiovasculares heredables es poder conocer qué familiares sin expresión fenotípica están en riesgo de desarrollar la enfermedad en un futuro. La detección de una mutación en una familia nos permite dar el alta definitiva a los familiares sin mutación y seleccionar para seguimiento clínico a aquellos con mutación. Sin embargo, es preciso hacer saber a las familias que el estudio genético presenta una serie de limitaciones. La primera es que su rentabilidad diagnóstica no es del 100%, pues varía desde el 25-40% para patologías como el síndrome de Brugada al 75-80% para el síndrome del QT largo1. Esto significa que en un porcentaje de casos el estudio no va a ofrecer ningún resultado. Dado el rápido avance de las técnicas diagnósticas y del descubrimiento de nuevos genes implicados, en estas familias con resultado negativo debería ser plausible el planteamiento futuro de una nueva búsqueda de mutaciones, para lo que la disponibilidad de biobancos donde guardar las muestras de ADN sería deseable. Una segunda limitación del estudio genético, ya señalada, es el hallazgo de variantes con significado patogénico incierto. Este tipo de hallazgo se produce cada vez con mayor frecuencia a medida que la práctica de la NGS de amplios paneles con múltiples genes se está extendiendo. El consejo genético aquí ha de ser muy cauto, explicando a la familia las limitaciones del resultado obtenido.
La atención a los pacientes y familias con cardiopatías hereditarias idealmente ha de realizarse en unidades específicas especializadas, formadas por equipos multidisciplinares compuestos por cardiólogos expertos en este tipo de patología, genetistas, psicólogos y enfermería especializada.
Conflicto de interesesNo existe conflicto de intereses.
A Julia López Buiza por su colaboración en el diseño de las figuras.

