



La enfermedad de Tangier es una dislipemia genética infrecuente con herencia autosómica recesiva, producida por mutaciones del gen que codifica ABCA1. En ausencia de este transportador de colesterol, acontece una acumulación lipídica patológica que genera una auténtica tesaurismosis de colesterol. A nivel cardiovascular, es bien conocida la asociación de este síndrome con una aterosclerosis precoz. Además, el depósito de colesterol también puede alterar la estructura y función valvular, con gravedad y repercusión clínica variables. Exponemos un caso de enfermedad de Tangier con afectación cardiovascular extensa en forma de cardiopatía isquémica y valvular.
Tangier disease is a rare inherited autosomal recessive genetic dyslipaemia, caused by mutations of the gene that codes ABCA1. In the absence of this cholesterol transporter, there is a pathological accumulation of lipids which creates an authentic cholesterol thesaurismosis. At cardiovascular level, the association of this syndrome with early atherosclerosis is well known. Furthermore, cholesterol deposits can alter valve structure and function, with varying clinical severity and repercussions. We present a case of Tangier Disease with extensive cardiovascular involvement in the form of an ischaemic heart and valve disease.
La enfermedad de Tangier es un síndrome de deficiencia severa de HDL-C con riesgo de aterosclerosis precoz. La acumulación patológica de colesterol en diversos órganos permite clasificarla como enfermedad de depósito. A continuación presentamos un ejemplo de enfermedad de Tangier y sus posibles consecuencias.
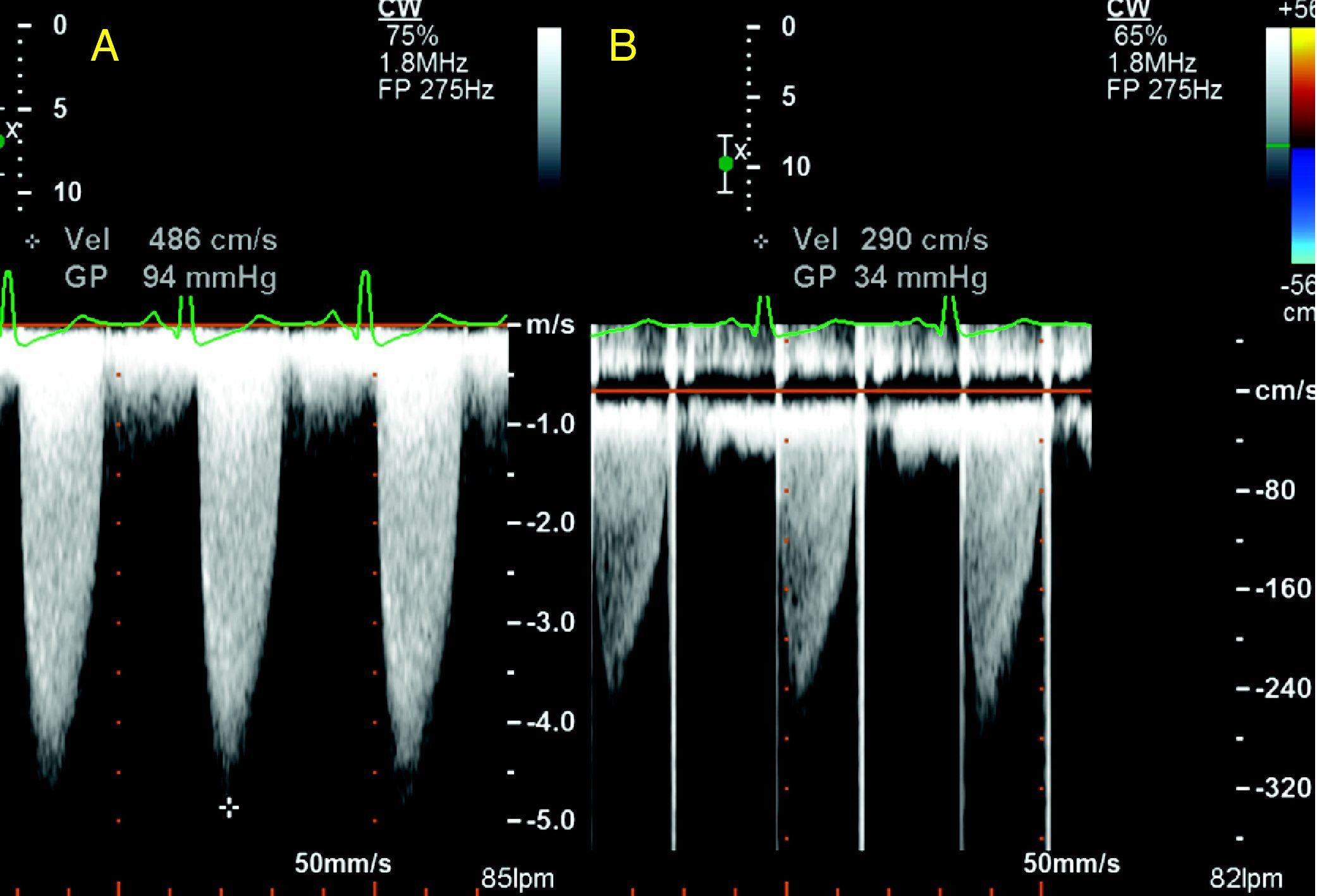
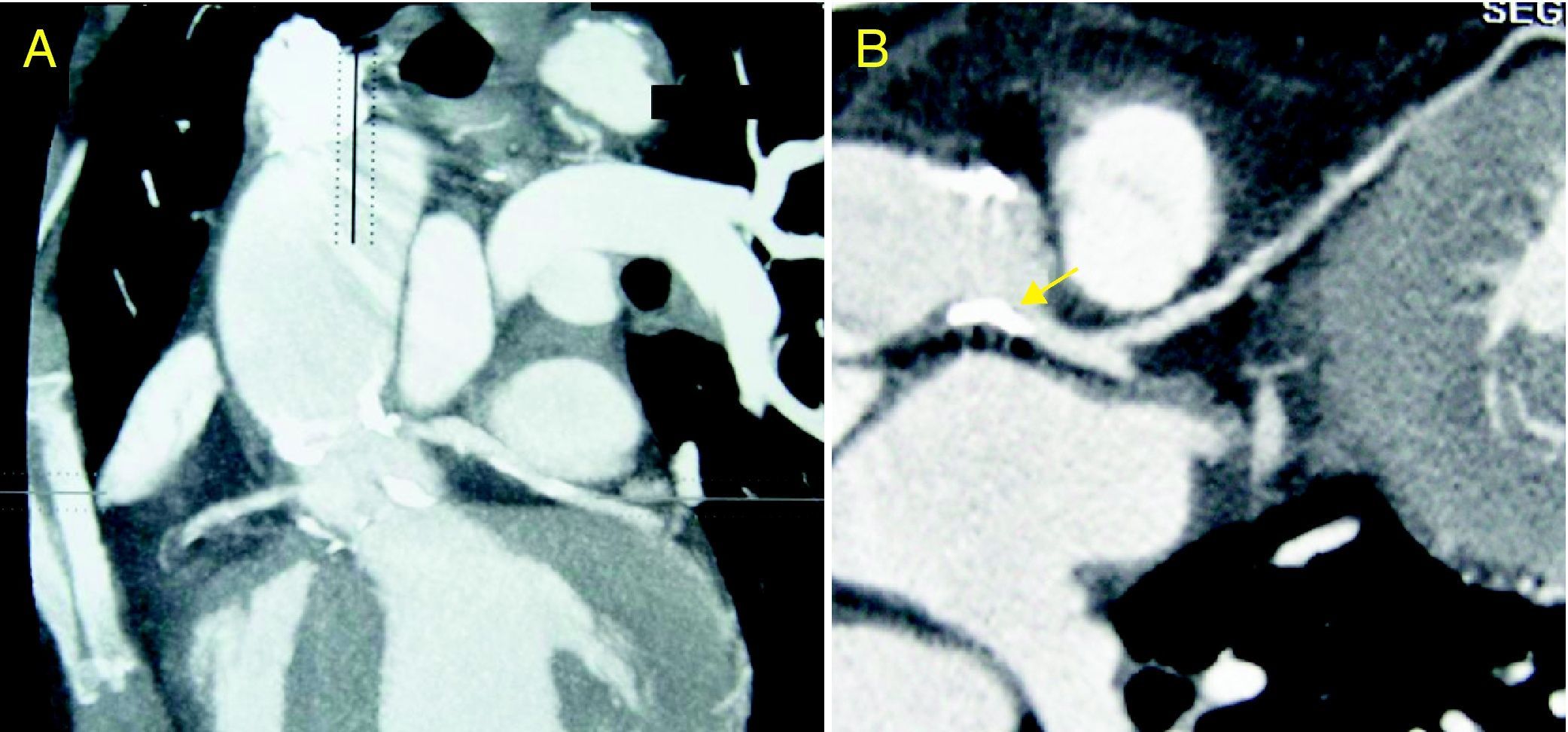
Mujer de 47 años remitida para estudio de disnea y angina de esfuerzo. Destacaba entre sus antecedentes personales amigdalectomía y adenoidectomía en la infancia por hipertrofia glandular sintomática (obstrucción nasal, infecciones recidivantes). El año previo había sido esplenectomizada por hiperesplenismo. A la exploración física destacaba soplo eyectivo aórtico III/VI con segundo ruido ausente. Los voltajes electrocardiográficos cumplían criterios de hipertrofia ventricular izquierda. La sospecha de valvulopatía aórtica fue confirmada por ecocardiograma: válvula aórtica trivalva, con calcificación de velos y apertura severamente reducida (área 0,45 cm2; gradiente transvalvular máximo de 94 mmHg) (fig. 1A), función sistólica normal. La bioquímica mostró un perfil lipídico alterado (colesterol total, 48 mg/dl; HDL-C, 3 mg/dl, LDL-C, 13 mg/dl; triglicéridos, 330 mg/dl; ApoA, 7 mg/dl [108-225]; ApoB, 125 mg/dl [60-117]). Se realizó una angio-TC torácica para valorar la aorta torácica y las arterias coronarias, evidenciando importante calcificación valvular, dilatación postestenótica de la raíz y afectación ateromatosa de la aorta ascendente (fig. 2A). El hallazgo de calcificación de la raíz aórtica y el tronco coronario izquierdo (fig. 2B) obligó realizar una coronariografía. Se encontraron lesiones en el tercio medio de la coronaria derecha y el ostium del tronco coronario. Con ello, se programó de forma preferente para cirugía de recambio valvular (prótesis mecánica), sustitución parcial de aorta ascendente y revascularización coronaria (puentes de mamaria interna a descendente anterior y safena a coronaria derecha). En el ecocardiograma postoperatorio el gradiente transprotésico máximo fue de 34 mmHg, medio de 20 mmHg (fig. 1B), con fracción de eyección normal. El tratamiento al alta incluyó anticoagulación oral, bloqueadores beta, IECA y fibratos (estatinas no toleradas por mialgias; hipertrigliceridemia predominante). En el seguimiento cardiológico al año, la paciente mantiene grado funcional I para angina y disnea. Realiza inmunoprofilaxis contra gérmenes encapsulados y de endocarditis infecciosa. Destaca la reciente adición de gabapentina debido a la aparición de dolor neuropático secundario a neuropatía periférica.


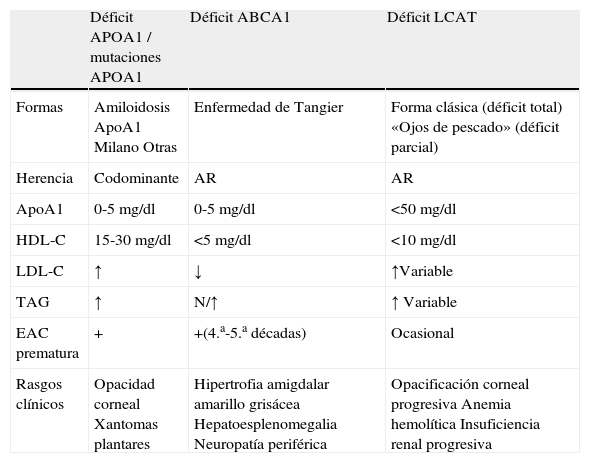
La enfermedad de Tangier es un dislipemia genética infrecuente con herencia autosómica recesiva, producida por mutaciones del gen que codifica ABCA1. En ausencia de este transportador celular encargado del transporte reverso de colesterol, acontece una acumulación lipídica patológica en fibroblastos y macrófagos1. La presencia de amígdalas color anaranjado es un hallazgo prácticamente patognomónico de la enfermedad. Sin embargo, es frecuente que las amígdalas hayan sido extirpadas en el momento de la evaluación. La acumulación de lípidos en las vainas nerviosas es responsable de la clínica neurológica, en forma de neuropatías transitorias pero recidivantes. Los niveles de colesterol en plasma están por debajo de 120 mg/ml, con triglicéridos normales o ligeramente elevados. Reducciones importantes de colesterol HDL, apoA-I y apoA-II (<2-5% del normal) forman el perfil lipídico que identifica a los pacientes con enfermedad de Tangier, confirmando el diagnóstico2. La enfermedad de Tangier se asocia con un incremento de riesgo de enfermedad aterosclerótica prematura, aunque menor del que cabría esperar por los bajísimos niveles de HDL-C y apoA-I, y que se atribuye a una protección parcial por los niveles también descendidos de LDL-C3. La identificación de mutaciones en el gen ABCA1 refuerza este diagnóstico, pero su ausencia no lo excluye, dada su gran variabilidad genética (más de 100 mutaciones descritas)4,5. El conocimiento del papel de ABCA1 en la enfermedad de Tangier ha permitido investigar nuevos candidatos para manipular la progresión de la aterosclerosis6. Sin embargo, el tratamiento con hipolipemiantes suele ser ineficaz, y por tanto las medidas terapéuticas son puramente sintomáticas: exéresis paliativa de órganos infiltrados (en ocasiones antes de un diagnóstico cierto), revascularización coronaria según las recomendaciones para la población general. Existen varias formas monogénicas responsables de síndromes de déficit de HDL causadas por alteración de genes implicados en el trasporte reverso del colesterol. Si bien el patrón oro es la identificación del gen alterado, la aproximación clínico-analítica permite un diagnóstico diferencial de gran valor (tabla 1). El componente hereditario de estos síndromes exige un cribado en familiares de primer grado, basado en determinaciones del perfil lipídico. Nuestra paciente no tenía descendencia, y su único familiar de primer grado (padre) tenía un perfil lipídico normal (madre fallecida en periparto, datos no disponibles).
Diagnóstico diferencial: causas hereditarias de HDL bajo
| Déficit APOA1 / mutaciones APOA1 | Déficit ABCA1 | Déficit LCAT | |
| Formas | Amiloidosis ApoA1 Milano Otras | Enfermedad de Tangier | Forma clásica (déficit total) «Ojos de pescado» (déficit parcial) |
| Herencia | Codominante | AR | AR |
| ApoA1 | 0-5 mg/dl | 0-5 mg/dl | <50 mg/dl |
| HDL-C | 15-30 mg/dl | <5 mg/dl | <10 mg/dl |
| LDL-C | ↑ | ↓ | ↑Variable |
| TAG | ↑ | N/↑ | ↑ Variable |
| EAC prematura | + | +(4.a-5.a décadas) | Ocasional |
| Rasgos clínicos | Opacidad corneal Xantomas plantares | Hipertrofia amigdalar amarillo grisácea Hepatoesplenomegalia Neuropatía periférica | Opacificación corneal progresiva Anemia hemolítica Insuficiencia renal progresiva |
AR: autosómica recesiva; EAC: enfermedad arterial coronaria.
Otro dato a destacar es la necesidad de prevención de infecciones. Por un lado, la esplenectomía expone a infecciones por gérmenes encapsulados, estando indicada la vacunación contra Streptococcus pneumoniae, Neisseria meningitidis y Haemophilus influenzae tipo b. Por otro lado, la afectación valvular exige considerar profilaxis de endocarditis infecciosa7. Finalmente, determinados estudios han identificado proteínas relacionadas con el HDL-C con papel en la inmunidad innata, regulación del complemento e inflamación; aunque se desconoce su trascendencia en la enfermedad de Tangier, el descenso de niveles de HDL-C podría exponer a un riesgo aumentado de infecciones y cáncer8.

