



Presentamos un caso clínico de miocardiopatía hipertrófica obstructiva asociada a una variante rara de neurofibromatosis tipo 1, como es la neurofibromatosis espinal aislada múltiple, cuya asociación se ha reconocido excepcionalmente.
We report a case of hypertrophic obstructive cardiomyopathy associated with an unusual variant form of neurofibromatosis type 1: multiple isolated spinal neurofibromatosis.
La neurofibromatosis tipo 1 (NF1) o enfermedad de Von Recklinghausen es un trastorno neurocutáneo que puede afectar a otros sistemas como el esquelético, el ocular y —menos— el cardiovascular. Este último se ha descrito asociado con la miocardiopatía hipertrófica obstructiva (MCHO)1,2. La NF1 tiene una prevalencia de 1/3.000 nacidos. Es una enfermedad autosómica dominante, siendo en un 50% de los casos de aparición esporádica por mutación del gen NF1, localizado en el cromosoma 17q11.2, el cual codifica una proteína (neurofibromina) que actúa como reguladora negativa del oncogén ras3,4. Esta proteína se expresa en tejidos derivados de la cresta neural, y al menos 2 isoformas se detectan en el músculo cardíaco. El déficit de neurofibromina favorecería la aparición de tumores y la desorganización y el hipercrecimiento de la musculatura cardíaca en desarrollo.
Dada la excepcionalidad de esta asociación, creemos de interés la descripción de este caso.
Presentamos un paciente varón de 32 años, sin antecedentes familiares de muerte súbita, que fue diagnosticado con 13 años de MCHO septal asimétrica e insuficiencia mitral moderada, sin historia sincopal y con múltiples episodios de fibrilación auricular persistentes (FAP), mal toleradas. Destacaba un cuadro dismórfico consistente en pectus excavatum, implantación anómala de quinto dedo de ambos pies, escoliosis e hiperlaxitud ligamentosa junto con otras anomalías tales como disfunción plaquetaria y anemia ferropénica. Así mismo se recogió entre sus antecedentes la presencia de endocarditis bacteriana subaguda sobre válvula mitral en 1994 por estreptococo betahemolítico, resuelta con antibioticoterapia, e insuficiencia mitral moderada residual; un hematoma tálamo-mesencefálico derecho en 1999, con hidrocefalia obstructiva que precisó implantación de derivación ventrículo-peritoneal y hemiplejía izquierda residual y un SAOS secundario a hipotiroidismo por amiodarona que remitió tras su suspensión y tratamiento hormonal.
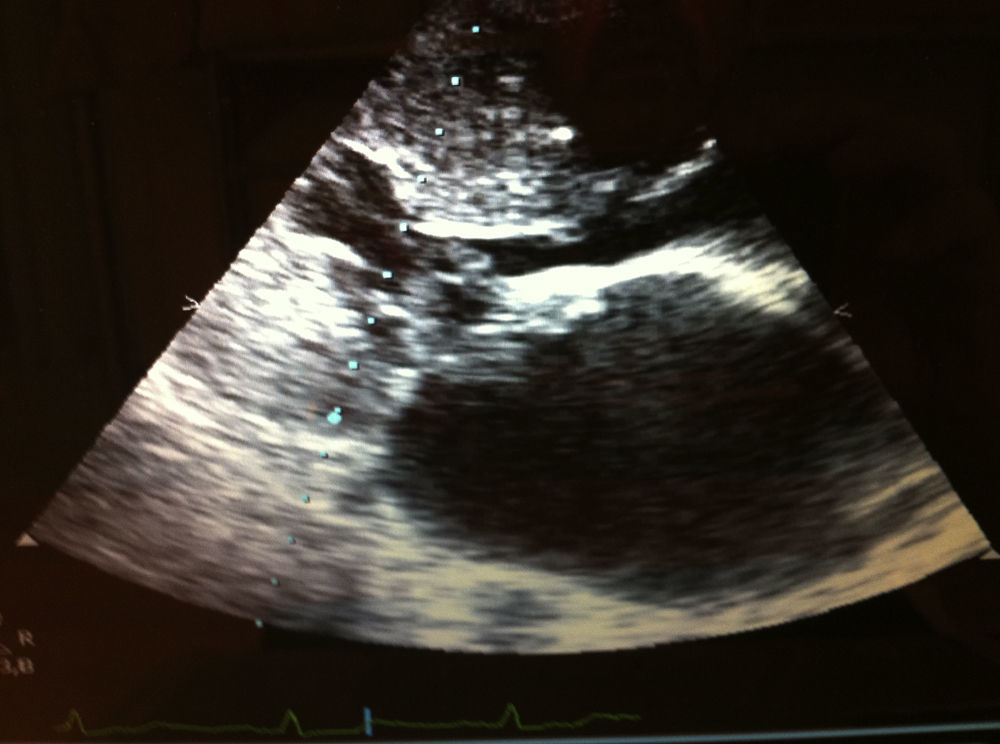
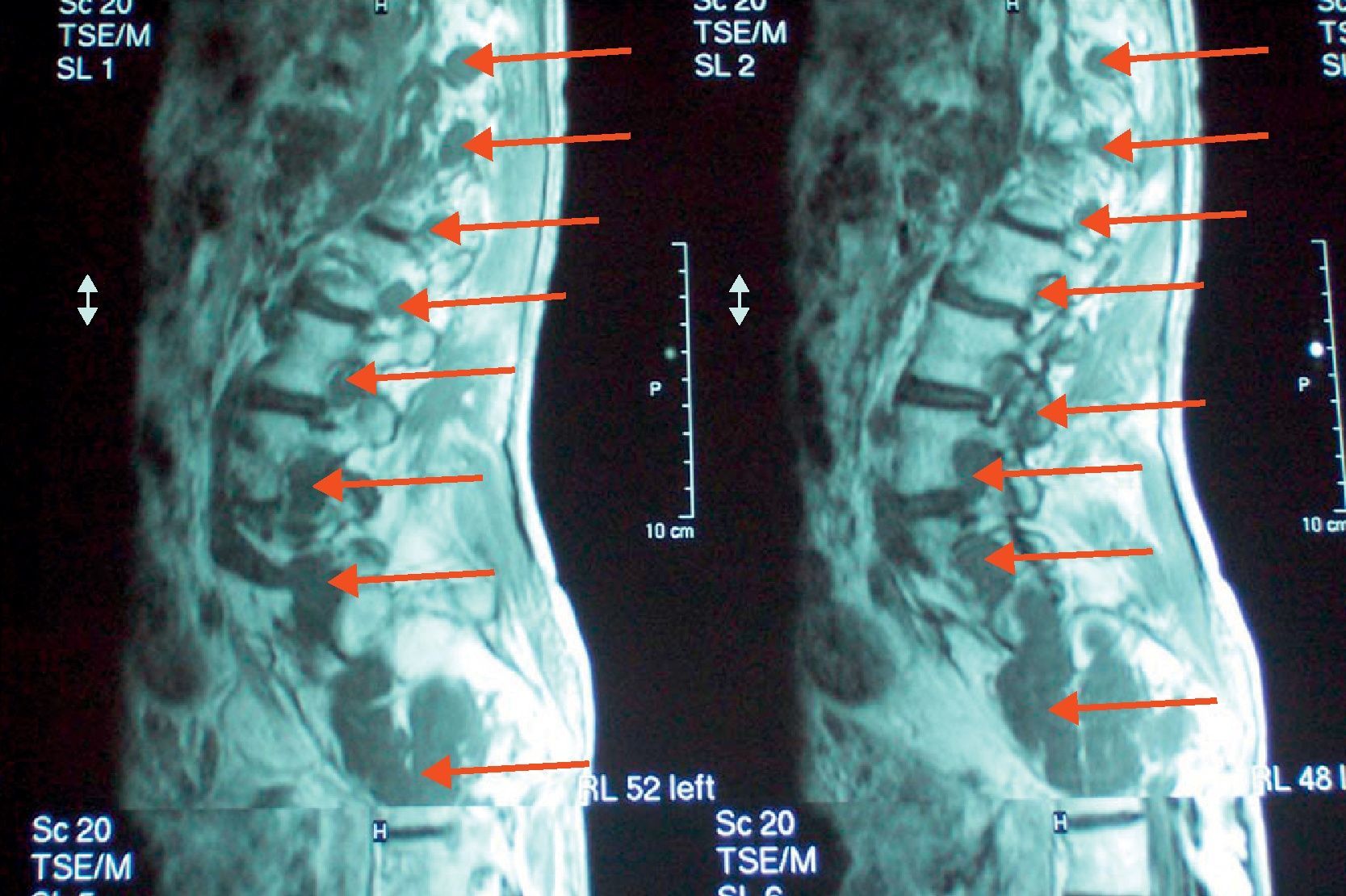
Ingresa por disnea progresiva y episodios de palpitaciones. En la exploración destacaban los hallazgos dismórficos mencionados y la presencia de un soplo sistólico en el ápex con disminución del primer tono y presión arterial de 100/60mmHg. El electrocardiograma mostró un ritmo sinusal y signos de hipertrofia ventricular izquierda. En la ecocardiografía-doppler destacaba una aurícula izquierda dilatada, la válvula mitral era displásica y desestructurada, con ligero prolapso del velo anterior con movimiento sistólico anterior del mismo, una severa hipertrofia de predominio septal (25mm) con pared posterior de 14mm y obstrucción del TSVI. El ventrículo izquierdo no estaba dilatado, con una fracción de eyección del 70%, y las cámaras derechas eran normales (fig. 1). El doppler mostró una insuficiencia mitral severa y gradiente dinámico en TSVI de 39mmHg sin hipertensión pulmonar. Holter-ECG: ausencia de TVNS. El estudio de imagen de aorta torácica y abdominal fue normal, con dilatación del tronco de la arteria pulmonar (45mm) y de ambas ramas. El estudio oftalmológico no mostró anomalías. La RMN de columna mostró múltiples lesiones dependientes de las raíces bilaterales, sobre todo a nivel dorsal bajo hasta S5, compatible con neurofibromas (fig. 2). Se realizaron ecocardiografías a sus padres y al único hermano, que resultaron ser normales. No se pudo realizar estudio genético por falta de accesibilidad.

.")
La evolución clínica cursó con múltiples episodios de FAP sin control adecuado de la frecuencia, optándose por implantación de marcapasos definitivo bicameral y ablación del nodo AV.
La alta comorbilidad del paciente y la no aceptación de la cirugía de la válvula mitral, declinaron el tratamiento quirúrgico. No se indicó DAI como prevención primaria de muerte súbita, ya que no reunía la presencia de al menos 2 criterios de riesgo admitidos (historia familiar de muerte súbita, síncope de origen no explicado, hipertrofia severa ≥30mm, múltiples episodios de TVNS en el holter-ECG y respuesta hipotensora en el test de ejercicio). El paciente, a los 2 años de su alta domiciliaria, permanece asintomático.
Nuestro paciente cumplía criterios de una variante rara de NF1 (neurofibromatosis espinal aislada múltiple)3 en la que faltan las otras manifestaciones oculocutáneas (nódulos de Lisch y manchas café con leche) y la afectación familiar. Su asociación con MCHO es muy variable según la literatura publicada, habiéndose informado en 27 pacientes2,5.
En la actualidad la neurofibromatosis forma parte de los síndromes neuro-cardio-facio-cutáneos, agrupando un conjunto de enfermedades hereditarias que pueden incluir afecciones como retraso del crecimiento y mental, dismorfia craneofacial, anomalías cardiacas (estenosis pulmonar y MH), predisposición a enfermedades hematológicas, tumores óseos y anomalías cutáneas. Se incluyen además los síndromes de Leopard, de Noonan y el cardio-facio-cutáneo. Parece que el nexo común entre ellos es que a pesar de ser debidos a distintas mutaciones genéticas, todas ellas interfieren en la regulación de las cinasas RAS-MAP, una cascada de transducción que regula la proliferación, diferenciación y supervivencia celular y que está implicada en la oncogénesis humana.
Así pues, parece prudente aconsejar estudio ecocardiográfico a los paciente con NF1, dada su asociación potencial.

