El origen anómalo del árbol coronario izquierdo de la arteria pulmonar (ALCAPA), también conocido como síndrome de Bland-White-Garland, es una cardiopatía congénita infrecuente. Aproximadamente el 90% de los pacientes fallecen en el primer año de vida.
Presentamos el excepcional caso de un varón de 79 años con dolor torácico, que fue diagnosticado mediante una tomografía computarizada coronaria de síndrome de Bland-White-Garland.
Origin of the anomalous left coronary artery from the pulmonary artery (ALCAPA), also known as Bland-White-Garland syndrome, is an uncommon congenital heart disease. Approximately 90% of patients die within the first year of life.
We report an exceptional case of a 79 year-old man, with chest pain who was diagnosed by multidetector computed tomography with Bland-White-Garland syndrome.
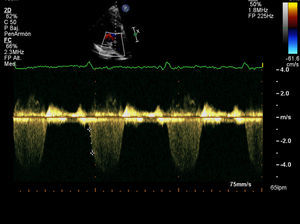
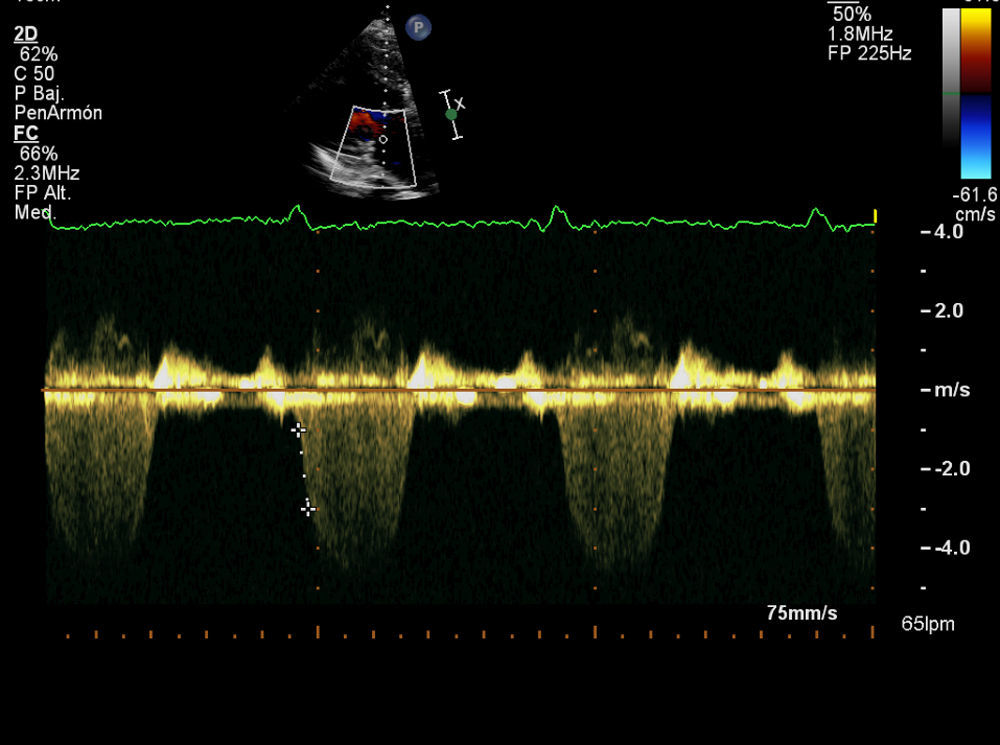
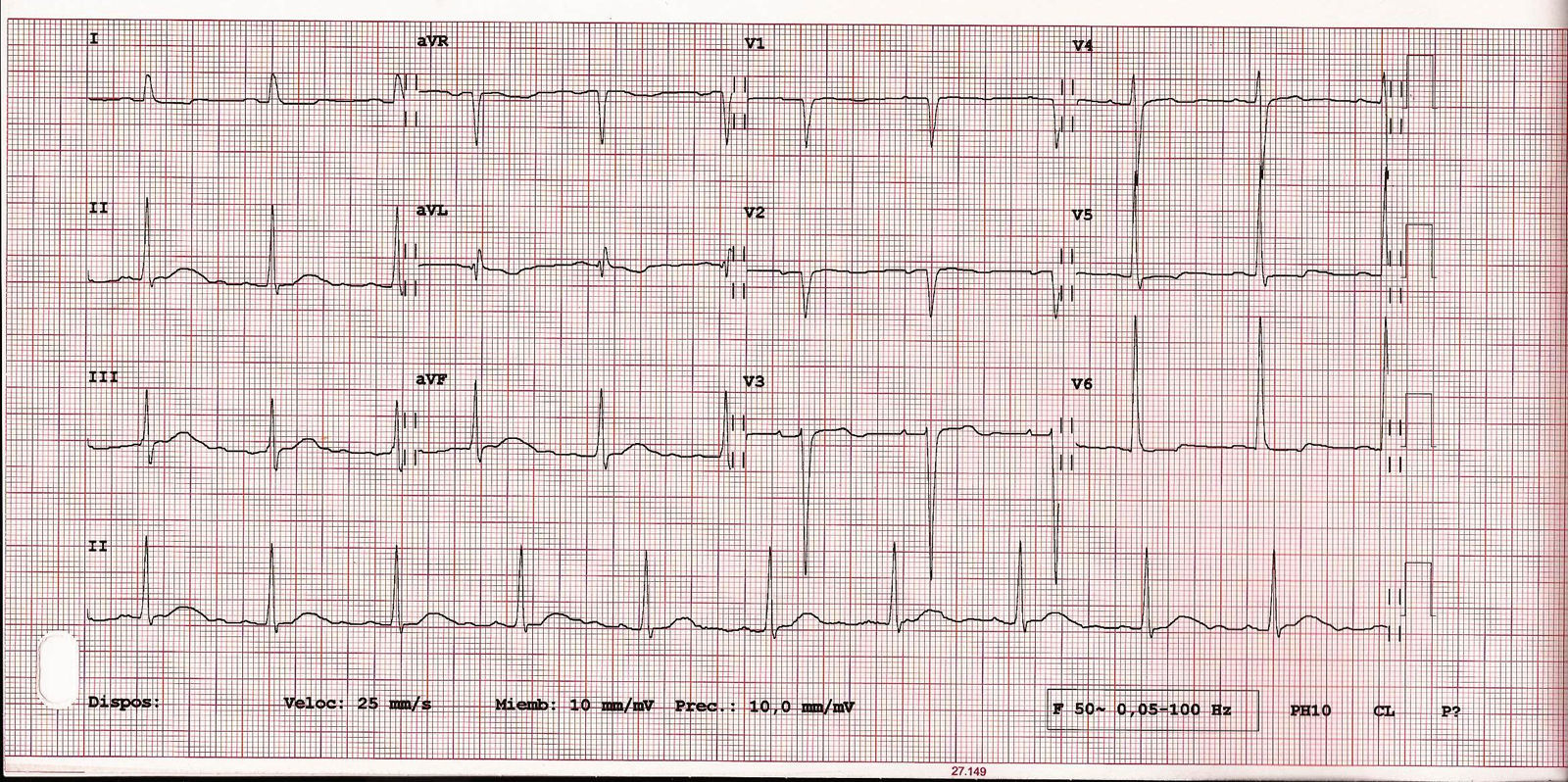
Varón de 79 años hipertenso, dislipémico y diabético. Consulta por angina a moderados esfuerzos de un año de evolución y palpitaciones ocasionales. En la auscultación destacaba un soplo continuo paraesternal derecho. El ecocardiograma reveló un situs inversus con concordancia aurículo-ventricular y ventrículo-arterial con un ventrículo izquierdo no dilatado, destacando una hipertrofia severa con septo de 20mm y señal de flujos intramiocárdicos por Doppler-color a este nivel. No se apreciaban alteraciones de la contractilidad segmentaria estando la función sistólica global conservada. Así mismo, presentaba una insuficiencia mitral degenerativa en grado ligero-moderado. El electrocardiograma estaba en ritmo sinusal con QS en derivaciones V1-V2 con onda T negativa en la cara lateral y lateral alta. Se realizó monitorización Holter 24h objetivándose extrasístoles ventriculares aisladas de moderada densidad sin arritmias sostenidas.
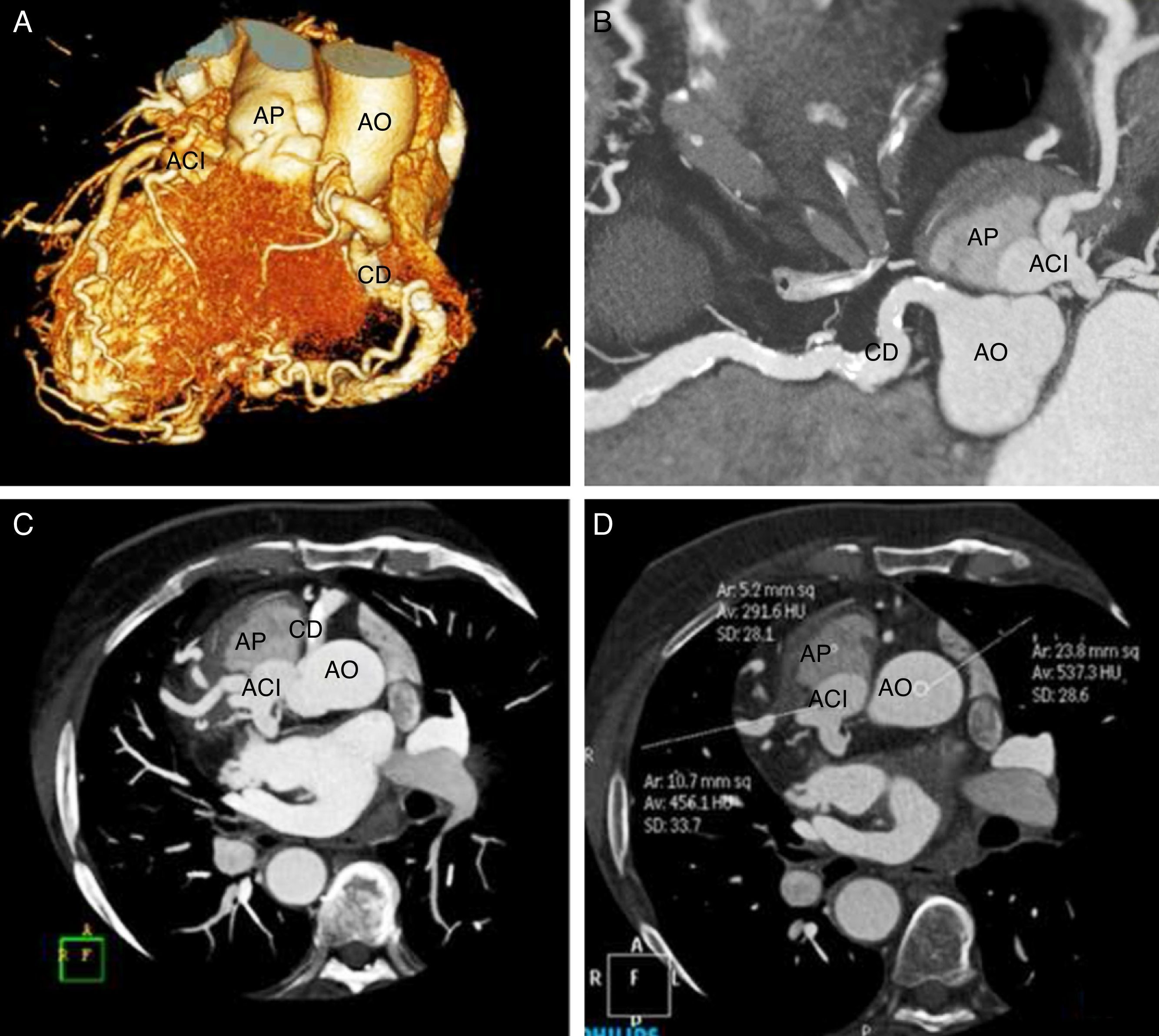
Se realizó una tomografía computarizada cardiaca (TCc) que mostró situs inversus con coronaria derecha (CD) de origen normal y trayecto ectásico con placas fibrocalcificadas sin estenosis luminal significativa. Sin embargo, el árbol coronario izquierdo (ACI) se originaba en la arteria pulmonar (AP), presentado una amplia red de colaterales entre ambos sistemas (fig. 1). El análisis de densidad (fig. 1D) sugería la existencia de un cortocircuito izquierda-derecha con flujo desde CD a ACI.
 y corte axial (C) en las que se observa la CD con origen normal y el Tronco Común Izquierdo originado en la Arteria Pulmonar. D: análisis del grado de atenuación en Unidades Hounsfield en el lumen de la Arteria Pulmonar, la Aorta y el Tronco Coronario Izquierdo. Obsérvese que las mediciones en la AO y en el ACI son prácticamente similares, lo cual sugiere la existencia de un cortocircuito izquierda-derecha. ACI: árbol coronario izquierdo; AO: aorta; AP: arteria pulmonar; CD: coronaria derecha.")
A: reconstrucción volumétrica en la que se observa la CD de gran tamaño con una amplia red de colaterales que van hacia el árbol coronario izquierdo. B y C: reconstrucción multiplanar (B) y corte axial (C) en las que se observa la CD con origen normal y el Tronco Común Izquierdo originado en la Arteria Pulmonar. D: análisis del grado de atenuación en Unidades Hounsfield en el lumen de la Arteria Pulmonar, la Aorta y el Tronco Coronario Izquierdo. Obsérvese que las mediciones en la AO y en el ACI son prácticamente similares, lo cual sugiere la existencia de un cortocircuito izquierda-derecha.
ACI: árbol coronario izquierdo; AO: aorta; AP: arteria pulmonar; CD: coronaria derecha.
Por la avanzada edad del paciente, así como un adecuado control sintomático con medicación, se desestimó el tratamiento quirúrgico optándose por un tratamiento conservador. Un año después, el paciente permanece con angina a moderados esfuerzos y no ha precisado ingreso hospitalario durante el seguimiento.
DiscusiónLa anomalía del origen del ACI en la AP, conocida como síndrome de Bland-White-Garland o anomalous origin of the left coronary artery from the pulmonary artery (ALCAPA), es una cardiopatía congénita infrecuente (uno de cada 300.000 recién nacidos vivos)1,2. Durante la vida fetal, puede ser bien tolerado gracias a que la presión en la AP es igual a la presión sistémica, por lo que el flujo es anterógrado tanto en el ACI anómalo como en la CD. Tras el nacimiento, cuando la presión en la AP disminuye, el flujo a través del ACI es menor y finalmente acaba invirtiéndose, generando hipoperfusión e isquemia miocárdica3. Debido a esto, el 90% de los afectados fallecen en el primer año de vida1,3. Sin corrección quirúrgica, menos del 10% alcanzan la edad adulta y los escasos pacientes que sobreviven, lo hacen gracias a la formación de numerosas colaterales entre la CD y el ACI que aportan una irrigación suplementaria al miocardio3. Las manifestaciones clínicas varían según el desarrollo de las colaterales. Con frecuencia aparece disfunción ventricular tardía e insuficiencia mitral secundaria a dilatación ventricular e isquemia de los músculos papilares2. Debido a la isquemia, se produce fibrosis subendocárdica que puede ser el sustrato para arritmias ventriculares y muerte súbita.
Hasta la aparición de las nuevas técnicas de imagen, la coronariografía era necesaria para llegar al diagnóstico de este síndrome. Sin embargo, el desarrollo de la TCc y la resonancia magnética permiten llegar al diagnóstico de una forma no invasiva4.
Los hallazgos ecocardiográficos incluyen una CD dilatada y la detección del ACI originándose en la AP, con abundantes colaterales septales que pueden observarse al realizar un estudio Doppler-color sobre el tabique interventricular4,5. En nuestro caso, aunque detectamos la presencia de abundantes colaterales septales, la TCc fue clave para identificar el origen y el trayecto de las coronarias. En el electrocardiograma es frecuente hallar ondas Q en las derivaciones de la cara anterior y alteraciones del segmento ST-T.
Su tratamiento definitivo es quirúrgico, habiéndose utilizado diversas técnicas: ligadura del tronco, anastomosis con la arteria subclavia, reimplante directo de la coronaria anómala en la aorta o el procedimiento de Takeuchi, consistiendo este último en la creación de una ventana aorto-pulmonar y un túnel intrapulmonar que conecta el ostium del tronco con la aorta2.
En nuestro caso, debido a la avanzada edad del paciente, se optó por un manejo conservador.