










El síndrome de Laugier-Hunziker es una hiperpigmentación macular mucocutánea benigna que afecta principalmente a mucosa bucal y uñas. Se presenta una serie de 6 casos, incluyendo 2 pacientes de una misma familia, se revisa la literatura y se discute su diagnóstico diferencial con otras enfermedades locales y sistmicas trascendentes que también muestran pigmentaciones maculares cutáneas, en labios y mucosa bucal, haciendo énfasis en las características clínicas como clave para su diagnóstico y tratamiento adecuado.
Laugier-Hunziker syndrome is a benign mucocutaneous pigmentary disorder that mainly affects the oral mucosa and nails. A series of six cases is presented, including two patients from the same family. A review of the literature is performed, as well as a discussion on its differential diagnosis from other relevant local and systemic pigmentary disorders with the same macular image. Emphasis is placed on the key clinical features for the diagnosis and proper treatment of this disease that affects the skin, lips and oral mucosa.
El síndrome de Laugier-Hunziker (SLH) es un trastorno benigno no hereditario, caracterizado por la presencia de múltiples máculas de color café oscuro a negro en mucosa bucal y labios, frecuentemente asociadas con melanoniquia longitudinal en ausencia de enfermedad sistémica1. La importancia clínica de esta condición radica en la necesidad de diferenciarla de otras enfermedades que presentan un cuadro clínico similar, las cuales pueden asociarse a compromiso sistémico diverso e incluso al desarrollo ulterior de neoplasias malignas. En este artículo se presenta una serie de 6 casos de esta rara afección mucocutánea en la que se describe el espectro clínico patológico y se discute su diagnóstico diferencial y conducta terapéutica.
Reporte de casosLa presente serie quedó integrada por 6 casos admitidos en los servicios de Clínica de Boca del Departamento de Dermatología del Hospital General “Dr. Manuel Gea González” y del Servicio de Medicina Bucal de las Clínicas Estomatológicas de la Universidad Autónoma Metropolitana Xochimilco en la ciudad de México, durante el período comprendido entre 2005 y 2013.
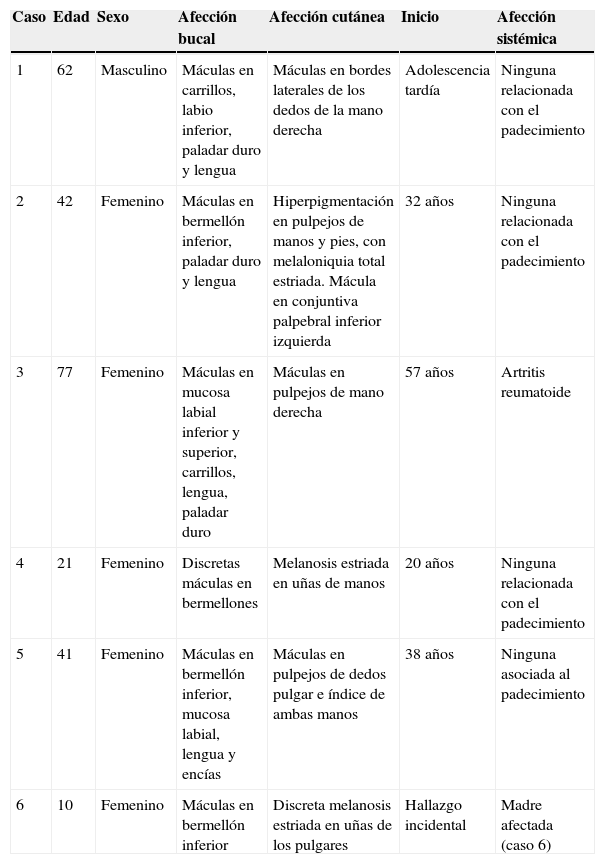
En la tabla 1 se presenta un resumen de las características clínicas y demográficas de los casos estudiados, observándose que hubo un hombre (16.6%) y 5 mujeres (83.3%), en un rango de edad de 10 a 77 años, y una media de 42.1. Las figuras 1-6 muestran los hallazgos clínicos sobresalientes. Se realizaron estudios histopatológicos en todos los casos, en los que se observó consistentemente la presencia de áreas de melanosis focal con caída de pigmento a la lámina propia superficial, sin alteraciones en número o forma de los melanocitos y sin datos de displasia epitelial (figs. 7 y 8).
Características clínicas y demográficas sobresalientes de 6 pacientes con síndrome de Laugier-Hunziker
| Caso | Edad | Sexo | Afección bucal | Afección cutánea | Inicio | Afección sistémica |
|---|---|---|---|---|---|---|
| 1 | 62 | Masculino | Máculas en carrillos, labio inferior, paladar duro y lengua | Máculas en bordes laterales de los dedos de la mano derecha | Adolescencia tardía | Ninguna relacionada con el padecimiento |
| 2 | 42 | Femenino | Máculas en bermellón inferior, paladar duro y lengua | Hiperpigmentación en pulpejos de manos y pies, con melaloniquia total estriada. Mácula en conjuntiva palpebral inferior izquierda | 32 años | Ninguna relacionada con el padecimiento |
| 3 | 77 | Femenino | Máculas en mucosa labial inferior y superior, carrillos, lengua, paladar duro | Máculas en pulpejos de mano derecha | 57 años | Artritis reumatoide |
| 4 | 21 | Femenino | Discretas máculas en bermellones | Melanosis estriada en uñas de manos | 20 años | Ninguna relacionada con el padecimiento |
| 5 | 41 | Femenino | Máculas en bermellón inferior, mucosa labial, lengua y encías | Máculas en pulpejos de dedos pulgar e índice de ambas manos | 38 años | Ninguna asociada al padecimiento |
| 6 | 10 | Femenino | Máculas en bermellón inferior | Discreta melanosis estriada en uñas de los pulgares | Hallazgo incidental | Madre afectada (caso 6) |




.")
Paciente 5. A. Lesiones en labio inferior. B. Lesiones pigmentadas en lengua, y C. Máculas en dedo (madre de la paciente de la Figura 6).
.")
.")
.")
Son diversas las entidades que se pueden manifestar con alteraciones en la pigmentación simultánea tanto en las mucosas como en la piel. Existe una gran variedad clínica en cuanto a la disposición y extensión de las máculas o manchas dependiendo del diagnóstico de base, y estas son usualmente de carácter benigno.
Los trastornos de la pigmentación bucal pueden deberse a la producción excesiva de melanina, o en algunos casos a la presencia de depósitos exógenos, tales como materiales de restauración dental (amalgama), medicamentos o la incrustación accidental de grafito, entre otros2. En ocasiones, pueden asociarse a enfermedades sistémicas o síndromes, lo cual debe ser considerado en el momento de realizar el diagnóstico diferencial.
La aparición espontánea de múltiples máculas hiperpigmentadas sobre la mucosa bucal y labios sin la presencia de enfermedad sistémica subyacente fue reportada en 1970 por Laugier y Hunziker, llamándole inicialmente “pigmentación melánica lenticular esencial de la mucosa bucal y labios”3. En 1979, Baran4 recalcó la importancia del hallazgo de la banda de melanoniquia longitudinal como una clave para el diagnóstico de SLH, al describir su presencia en 5 de 9 pacientes con esta condición.
Este síndrome se ha descrito con mayor frecuencia en personas caucásicas5,6, con predominio en mujeres, y muestra una prevalencia mayor en adultos jóvenes7. Su evolución se caracteriza por un aumento progresivo en el número de lesiones a través de los años8 hasta hacerse estable, como en la mayoría de los casos de esta serie. Hasta finales del año 2012 existían en la literatura internacional aproximadamente 172 reportes de casos de esta entidad9.
La causa de esta condición no se ha determinado y tampoco se ha descrito su asociación con otras enfermedades1. Makhoul et al.10 describieron un caso familiar de SLH, siendo este el único reportado a la fecha, por lo que resulta importante la inclusión en nuestra serie del caso de una niña de 10 años (caso 6) que fue identificada incidentalmente después de diagnosticar a la madre (caso 5) por la presencia de máculas labiales, gingivales, linguales y en dedos y uñas en ausencia de otras alteraciones sistémicas (figs. 5 y 6).
Pese a que este síndrome se ha calificado como una entidad benigna1, Simionescu et al. Reportaron, en el año 2008, el caso de una paciente afectada por SLH que presentó un melanoma en boca. Dicha paciente tenía una lesión pigmentaria de larga evolución (hiperplasia melanocítica) en la mucosa del labio superior izquierdo, misma que sufrió una progresión hacia un melanoma in situ y posteriormente a melanoma con invasión vertical11. Lo anterior establece la necesidad de realizar biopsia y un seguimiento cuidadoso de aquellos casos que presenten datos sugestivos de hiperplasia melanocítica y en los que se produzcan cambios en la evolución clínica de alguna de las lesiones que pudiesen sugerir transformación maligna. En nuestra serie no hubo hallazgos histopatológicos que sugieran alteraciones melanocíticas precursoras de malignidad, pero se informó la necesidad en cada caso de hacer biopsia y tener un seguimiento a largo plazo de las lesiones pigmentadas.
El SLH se caracteriza por la presencia de un número variable de máculas pigmentadas de color café oscuro a negro, asintomáticas, solitarias o confluentes, comúnmente lenticulares y a veces lineales1,9,12. Las zonas afectadas con mayor frecuencia incluyen la mucosa de carrillos, bermellones y piel de labios10, pero también se pueden observar en encías3,13, lengua14, comisuras15, faringe, esófago, nariz, conjuntiva, córnea9,13, vulva9,16–18, pene1,9 y dedos9,13. Es frecuente la asociación a melanoniquia longitudinal19, la cual se encuentra hasta en un 60% de los casos20.
Histológicamente, el SLH se caracteriza por acumulación de melanina en los queratinocitos basales, sin aumento de la población de melanocitos, y por un abundante número de melanófagos en la dermis superficial o corion9,10; sin embargo, en 2 reportes se ha señalado el hallazgo de un aumento en el número de melanocitos intraepidérmicos21,22, y en uno de ellos también se describió atipia celular significativa de los melanocitos intraepiteliales, en una lesión macular de un área fotoexpuesta21. Se ha descrito, además, la presencia de acantosis variable3,13,23 y elongación de papilas dérmicas22. Estos hallazgos son inespecíficos, por lo que tienen que ser considerados de manera conjunta con el cuadro clínico para establecer el diagnóstico1.
Entre los diagnósticos diferenciales de esta entidad se incluye principalmente al síndrome de Peutz-Jeghers (SPJ), la enfermedad de Addison, el melanoma24, el síndrome de LAMB o el complejo de Carney (léntigos en piel y mucosa bucal, mixomas auriculares y mucocutáneos, y múltiples nevos azules)25, síndrome de LEOPARD (léntigos, anormalidades electrocardiográficas, hipertelorismo ocular, estenosis pulmonar, anormalidades de genitales, retraso del crecimiento y sordera)26, entre otras entidades, por lo que es necesario realizar un examen detallado para diferenciarlo de estas27.
En el caso de SPJ, es importante destacar que las lesiones aparecen en la infancia temprana y presentan una distribución periorificial; las máculas y manchas de la piel pueden perder su color gradualmente; sin embargo, las lesiones intraorales tienden a persistir21, presentan una asociación frecuente a poliposis del tracto gastrointestinal que puede producir eventualmente dolor y obstrucción intestinal y los afectados poseen un riesgo incrementado para neoplasias10. El SPJ muestra una herencia autosómica dominante, el defecto se sitúa en el cromosoma 19 q13.3, pero hasta el 40% de los pacientes con esta enfermedad reportan antecedentes patológicos familiares negativos1. Se ha identificado el gen supresor de tumores STK11 (una serina-treonina quinasa), en las etapas más tempranas del desarrollo de hamartomas y adenocarcinomas en este síndrome28, por lo que se ha enfatizado la importancia de estudiar la alteración de este gen en pacientes con SLH, para determinar si está relacionado con el SPJ21.
La enfermedad de Addison resulta de la hipofunción de la corteza de la glándula suprarrenal, lo que provoca un aumento en la secreción de la hormona estimulante de la corteza suprarrenal y la hormona estimulante de melanocitos, lo que da lugar a las alteraciones pigmentarias en piel y mucosas que caracterizan a esta entidad8. Las zonas afectadas con más frecuencia son las áreas expuestas (dorso de manos, cara) y en regiones que presentan de manera fisiológica una mayor pigmentación (pliegues palmares, areolas, genitales externos, ano), además de cicatrices y mucosa oral. Solo de manera excepcional se ha reportado pigmentación en el aparato ungueal16. La apariencia clínica de las máculas pigmentadas del SLH se distinguen fácilmente de la pigmentación causada por la enfermedad de Addison, la cual es más acentuada y difusa21.
Es importante excluir los casos de pigmentación mucocutánea secundaria a condiciones extrínsecas, que podrían ser provocadas por tabaco2, medicamentos (por ejemplo: tetraciclinas, antimaláricos, amiodarona, quimioterapéuticos, anticonceptivos orales, zidovudina, ketoconazol, etc.)29 o por depósito de metales pesados (plata, oro, bismuto, mercurio)30.
En los casos confirmados de SLH, el tratamiento se considera meramente cosmético. Las opciones disponibles incluyen el láser de neodimio, itrio, aluminio y granate y Alexandrita Q-switched31–34. También se han reportado resultados efectivos con el tratamiento con criocirugía35. Es importante indicar medidas de protección solar en estos pacientes31, especialmente si recibieron alguno de los tratamientos mencionados36,37. Hay que recalcar que dichos recursos, que solo tienen fines cosméticos, podrían llevarse a cabo si el paciente está previamente informado y consiente en su realización.
Moore et al. han propuesto evitar el término “síndrome” para definir la alteración descrita inicialmente por Laugier y Hunziker, debido a que no se la ha podido asociar a otras anormalidades somáticas. Por este motivo, consideran más apropiado usar el término “pigmentación de Laugier y Hunziker” en lugar de “síndrome de Laugier-Hunziker”21. Al respecto, previamente se había utilizado también la denominación de pigmentación mucocutánea lenticular idiopática como sinónimo de SLH2,3,5
ConclusionesEl SLH es un diagnóstico al cual se llega por exclusión. La realización de una adecuada historia clínica es indispensable, y es importante tener en mente sus diagnósticos diferenciales debido a las afecciones sistémicas que podrían estar presentes si el diagnóstico fuese erróneo. En varias ocasiones serán el dermatólogo o el estomatólogo los primeros en constatar la enfermedad, por lo que es necesario que ambos especialistas tengan en consideración esta entidad, aunque no sea un diagnóstico frecuente en nuestra población. Respecto al manejo de las lesiones, debe ser considerado únicamente en casos severos con compromiso estético, y la toma de biopsia es necesaria para descartar lesiones potencialmente agresivas. La decisión terapéutica será tomada solo con fines terapéuticos mediante diversas opiniones.