




Systemic inflammation is characterised by high circulating levels of inflammatory cytokines and increased macrophage infiltration in peripheral tissues. Most importantly, this inflammatory state does not involve damage or loss of function of the infiltrated tissue, which is a distinctive feature of the low-grade systemic inflammation. The term “meta-inflammation” has also been used to refer to the low-grade systemic inflammation due to its strong relationship with the development of cardio-metabolic diseases in obesity.
ObjectiveA review is presented on the recent clinical and experimental evidence concerning the role of adipose tissue inflammation as a key mediator of low-grade systemic inflammation. Furthermore, the main molecular mechanisms involved in the inflammatory polarisation of macrophages with the ability to infiltrate both the adipose tissue and the vascular endothelium via activation of toll-like receptors by metabolic damage-associated molecular patterns, such as advanced glycation-end products and oxidised lipoproteins, is discussed. Finally, a review is made of the pathogenic mechanisms through which the low-grade systemic inflammation contributes to develop insulin resistance, dyslipidaemia, atherogenesis, type 2 diabetes, and hypertension in obese individuals.
ConclusionsA better understanding of the molecular mechanisms of low-grade systemic inflammation in promoting cardio-metabolic diseases is necessary, in order to further design novel anti-inflammatory therapies that take into consideration clinical data, as well as the circulating levels of cytokines, immune cells, and metabolic damage-associated molecular patterns in each patient.
La inflamación sistémica se caracteriza por la elevación en los niveles circulantes de citocinas inflamatorias; así como aumento en la infiltración de macrófagos en tejidos periféricos. Este escenario inflamatorio no induce lesión o pérdida de la funcionalidad en el tejido infiltrado, rasgo distintivo de un estado de inflamación sistémica de grado bajo. La inflamación sistémica de grado bajo posee una estrecha relación con el desarrollo de enfermedades cardiometabólicas en el paciente con obesidad, por lo que este estado de alteración inmune también ha recibido el nombre de metainflamación.
ObjetivoEn esta revisión presentamos la evidencia clínica y experimental más reciente en torno al papel de la inflamación del tejido adiposo como detonante de la metainflamación. Además, revisamos a nivel molecular los mecanismos de polarización inflamatoria de macrófagos invasores del tejido adiposo y el endotelio vascular a través de la activación de receptores tipo toll por patrones moleculares asociados a daño metabólico, tales como proteínas glucosiladas y lipoproteínas oxidadas. Por último, revisamos los mecanismos fisiopatogénicos de la inflamación sistémica en el desarrollo de resistencia a la insulina, dislipidemia, aterogénesis, diabetes mellitus tipo 2 e hipertensión en el paciente obeso.
ConclusionesUn entendimiento más detallado de los mecanismos moleculares a través de los cuales la inflamación sistémica de grado bajo promueve el desarrollo de enfermedades cardiometabólicas podría ser útil en el diseño de terapias antiinflamatorias que tengan en cuenta datos clínicos, así como el nivel circulante de citocinas, células inmunes y patrones moleculares asociados a daño metabólico en el paciente.
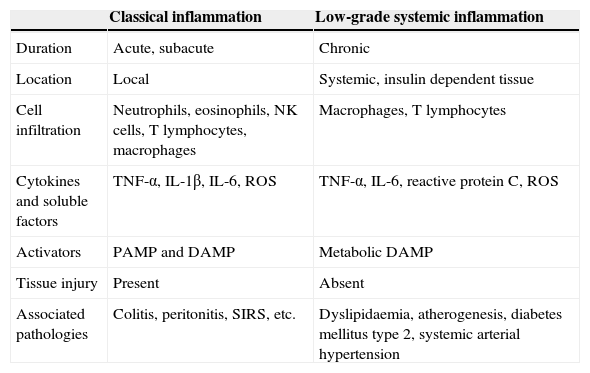
Recent experimental and clinical evidence suggests that metabolic syndrome and cardiovascular disease could be the consequence of a systemic inflammatory process.1 Systemic inflammation has major differences to a classic inflammatory response (Table 1). Specifically, systemic inflammation is characterised by high circulating levels of acute-phase proteins and active inflammatory cytokines, including C-reactive proteins (pCr), tumour necrosis factor alfa (TNF-α) and interleukins (IL) 1β, 6 and 17, in addition to an increase in immune cell infiltration including that of macrophages and t lymphocytes in insulin-dependent tissue.2,3 Furthermore, there is a consensus of opinion that systemic inflammation does not cause damage in immunologically infiltrated tissue; this distinctive trait has led to the term low-grade systemic inflammation.3,4 In other words, during a state of low-grade systemic inflammation tissues express high levels of inflammatory factors and immune cell infiltration whilst simultaneously not expressing any structural changes or loss of primary functions. By contrast, due to its close relationship with the development of cardio-metabolic diseases in obese patients, low-grade systemic inflammation has recently been referred to as meta-inflammation or metabolic inflammation.5
Main differences between classical and low-grade inflammation.
| Classical inflammation | Low-grade systemic inflammation | |
|---|---|---|
| Duration | Acute, subacute | Chronic |
| Location | Local | Systemic, insulin dependent tissue |
| Cell infiltration | Neutrophils, eosinophils, NK cells, T lymphocytes, macrophages | Macrophages, T lymphocytes |
| Cytokines and soluble factors | TNF-α, IL-1β, IL-6, ROS | TNF-α, IL-6, reactive protein C, ROS |
| Activators | PAMP and DAMP | Metabolic DAMP |
| Tissue injury | Present | Absent |
| Associated pathologies | Colitis, peritonitis, SIRS, etc. | Dyslipidaemia, atherogenesis, diabetes mellitus type 2, systemic arterial hypertension |
DAMP: damage associated molecular patterns; ROS: reactive oxygen species; NK: natural killer cells; IL: interleukin; PAMP: pathogen-associated molecular patterns; SIRS: systemic inflammatory response syndrome; TNF: tumour necrosis factor.
However, despite the fact that the relationship between low-grade systemic inflammation and cardio-metabolic diseases has considerably increased during the last decade, many elements are yet to be researched and discussed in this regard. For example, which cellular mechanisms drive the start and perpetuation of meta-inflammation? Which molecular events are involved in the clinical outset of systemic inflammation? Is there any existing evidence to suggest that low-grade systemic inflammation is the backdrop to pathologies such as insulin resistance, dyslipidaemia, high blood pressure and diabetes mellitus type 2?
Our approach in this review is to tackle each of the before-mentioned points, exposing the most recent information with regards to low-grade systemic inflammation and its relationship with the development of cardio-metabolic changes from a molecular and clinical viewpoint. Particular emphasis will be placed on the application of this knowledge to the identification of possible biomarkers and therapeutic targets which may lead to improvements in the treatment of patients with metabolic changes.
Inflammation of visceral adipose tissue: the beginning of low-grade systemic inflammationOne of the first mechanisms involved at the start of low-grade systemic inflammation is the inflammation of white or visceral adipose tissue (Fig. 1) As a consequence of the imbalance between energy intake and outputs, adipocytes tend to accumulate large amounts of fatty acids inside them, which leads to the white adipose tissue expanding in processes such as adipocitary hyperplasia and hypertrophy (increase in number and size, respectively). In response to modifications in the domain of the stromal vascular fractioning of the visceral adipose tissue, several adipocytes located in areas at some distance from blood vessels suffer from hypoxia and later from necrosis, after which they are surrounded by phagocytic cells which initiate an inflammatory state aimed at removing these cells.6 The large amount of fatty acids stored in these cells may exacerbate oxidative processes such as lipid peroxidation, consisting of the oxidation of lipid molecules inside the adipocyte. Lipid peroxidation which develops during adipocitary hyperplasia/hypertrophy induces a scenario of cellular oxidative stress characterised by a considerable rise in reactive species levels of oxygen and nitrogen, including superoxide ions (O2−) and nitric oxide (ON) respectively. A consequence of this oxidative explosion the recruitment of numerous immunological cells from the periphery to the adipose tissue, initiating a local level inflammatory process characterised by a rise in TNF-α and leptin levels, and a reduction in IL-10 and adiponectin.6 However, under stressful conditions such as hypoxia and hyperoxidation of fatty acids, functional changes occur in the adipocytes characterised by marked reticular stress associated with incorrect protein folding processes and autophagy, with possible onset of apoptosis in these cells.7 In addition to the cellular events described above, this apoptotoic process converges at an essential point in the initiation of the low-grade systemic inflammation, the inflammation of white adipose tissue.

Inflammation of visceral adipose tissue is a trigger signalling the beginning and the spread of low-grade systemic inflammation. AGE: advanced glycation end products; FFA: free fatty acids; IL: interleukin; LGSI: low-grade systemic inflammation; oxLDL: oxidised low density lipo proteins; TNF: tumour necrosis factor.
Histologically, the inflammation of adipose tissue is characterised by an infiltration mainly composed of macrophages and secondarily cytotoxic T lymphocytes, with the absence of or very little presence of neutrophils.3,8 The macrophages are surrounded by adipocytes forming a crown-like structure (CLS); a distinctive feature of low-grade inflammation in adipose tissue.6 It is important to underline that the infiltration of immune cells is, per se, a permanent source of cytokines and pro-inflammatory factors in hyperplasic and hypertrophic adipose tissue exhibiting high levels of IL-1β, IL-6, TNF-α and leptin, in addition to a greater production of proteins with chemo-attractive or chemokine proteins.9 Hyperplasic and hypertrophic adipose tissue express high concentrations of chemokines such as the macrophage chemo-attractive protein (MCP-1), the macrophage inhibitory factor (MIF-1) and RANTES, all of which are able to attract more macrophages and peripheral lymphocytes, thus perpetuating the tissue immune invasion process. Moreover, there is a growing body of evidence to suggest that changes on hyperplasic and hypertrophic visceral adipose tissue level changes may not only have autocrine and paracrine but also endocrine involvement, due to the fact that in patients with different obesity levels significantly higher elevated circulating levels of inflammatory cytokines, chemokines and macrophages and monocytes have been observed.1,2,10
During obesity inflammation of the visceral adipose tissue may therefore trigger a systemic inflammation process, which affects other insulin-dependent tissues such as those of the liver and muscles, thus perpetuating the inflammatory state on an adipocytary level. Up until now, we have described the cellular environment underlying visceral adipose tissue inflammation and its possible contribution to the development of low-grade systemic inflammation. But which molecular mechanisms are involved in the polarisation of the macrophage towards the inflammatory profile with its consequent release of TNF-α and the establishment of low-grade systemic inflammation? We will address this issue below.
The beginning of low-grade systemic inflammation depends on the activation of pattern recognition receptorsImmune cells, such as macrophages and dendritic cells express a group of membrane receptors which form part of the innate response to external and internal stimulae known as pattern recognition receptors (PRRs). In general, PRRs are able to detect two types of molecules, the pathogen-associated molecular patterns (PAMP) and the damage-associated molecular patterns (DAMP). PAMP are molecules derived from pathogen microorganisms whilst DAMP are released by tissues which have suffered some type of damage. The interaction between PRRs with PAMP or DAMP leads to rapid response to infection or damage to tissue respectively, through the activation of the inflammatory response and/or tissue repair. It is important to mention that the term PAMP is currently subject to debate since non-pathogen microorganisms also possess many of these molecular structures and are capable of inducing an immune response, such as, for example, that of immunological tolerance to normal intestinal microflora.11
Within the different PRR groups, the group made up of toll-like receptors (TLR) has been broadly characterised as a group of crucial receptors for recognition of both PAMP and DAMP.12 Ten functional types of TLR have been described in human beings, which may be located either on a cellular membrane level or cytoplasm level. On binding with ligands, the TLRs trigger an intracellular signalling cascade which culminates in the onset of an inflammatory response through the secretion of inflammatory and chemotactic mediators and the recruitment of immune cells to the infected or damaged cells. Although the role of TLR in infectious processes has been extensively described, a growing body of evidence suggests that these receptors could play a key role during the start and perpetuation of low-grade systemic inflammation associated with obesity.13
Unlike acute or classical inflammation induced by PAMP, the response associated with different molecules derived from a changed metabolic state or “metabolic DAMP” leads to a lower intensity, longer-lasting systemic inflammation.14 Different metabolites are linked as generators of low-grade systemic inflammation through their adhesion to TLRs.14 In this way, both dyslipidaemia and hyperglycaemia have shown to decisively participate in the polarisation of macrophages towards a pro-inflammatory phenotype and the concomitant initiation of low-grade systemic inflammation through activation of TLRs. Specifically, it has been known for some time that free fatty acids (FFA), and especially all the saturated ones such as palmitic acid, provoke a to systemic inflammatory response.12 At present we know that this phenomenon presents because during dyslipidaemia a large quantity of FFAs may adhere to TLR-1, 2 and 6 located in the surface area of a macrophage, forming heterodimers which join together with diacyl or triacy lipopeptides and release inflammatory cytokines.15
Moreover, apart from recognising lipopolysaccharride (LPS, a molecule derived from gram-negative bacteria), TLR-4 is also able to recognise glycated serum proteins, referred to as advanced glycation end products (AGE).16 During a hyperglycemic state nonenzymatic glycosylation takes place in several proteins including haemoglobin, albumin or low-density lipoproteins (LDL) in lysine and arginine residues giving rise to AGEs. In cells such as macrophages these AGEs may adhere to TLR-4, which is over expressed in metabolic diseases.13 Stimulation of TLRs imitates a signalling intracellular cascades mediated by the nuclear factor kappa B (NF-κB), which, once it has translocated to the nucleus, activates the transcription of genes encoding inflammatory cytokines and chemokines, including IL-1β, TNF-α and CXCL8, thereby initiating an inflammatory response in response to the hyperglycaemia.
Altered metabolic states such as dyslipidaemia and hyperglycaemia may therefore induce an inflammatory response in the macrophage through PRR activation, which could later spread systemically when this cell migrates towards insulin-dependent tissue and changes the microenvironment of cytokines in the body. These studies thus show that one of the possible therapeutic strategies for the control of low-grade systemic inflammation in the obese patient involves the design of drugs which would impede the adhesion of FFA and AGE to TLR-1, 2, 4 and 6, or which would block the activation of cellular mediators such as NF-κB, thereby potentially inhibiting the progress of systemic inflammation and its metabolic consequences. However, the clinical relevance of this type of new strategy is yet to be determined.
Our review of information up until now has enabled us to gain greater in-depth awareness of cellular and molecular mechanisms involved in the establishment of low-grade systemic inflammation during obesity. However, a couple of interesting questions arise for the remainder of this document. What are the clinical manifestations at the onset and for the duration of low-grade systemic inflammation in the obese patient? Is there any existing clinical evidence regarding the relationship between low-grade systemic inflammation and the development of atherogenesis, diabetes mellitus type 2 and systemic arterial hypertension? Our response to these questions is addressed below.
Dyslipidaemia, atherogenesis and low-grade systemic inflammationThere has recently been a rise in evidence to suggest that atherogenesis could be the consequence of a meta-inflammatory process with vascular involvement, in which the formation of a plaque partially or totally obstructs arterial light, leading to events such as myocardial infarction, cerebral vascular disease or sudden death.17 Both low-grade systemic inflammation and dyslipidaemia have been identified as decisive factors in atherogenesis and the vascular risks involved therein.17 In the nascent platelet, oxidised low density lipo proteins (oxLDL) adhere to receptors found on the macrophage surface, such as CD36, TLR-4, the receptor for advanced glycation end-products (RAGE) and the formyl-peptide receptor 2 (FPR2).18–20 This interaction initiates the release of ROSs, which activate the nucleotide-binding oligomerization domain-like receptor family of proteins (NLRP3), a complex of proteins with inflammatory and apoptotic activity in response to PAMP and DAMP.21 It has also been observed that the FFAs are able to directly release inflammatory agents (TNF-α and IL-1β) and oxidants (ON) in the endothelial cell when inflammasome-free mitochondrial uncoupling takes place.22 This cascade of inflammatory events causes the vascular endothelium to release adhesive molecules including the intracellular adhesion molecule 1 (ICAM-1) and the vascular cellular adhesion molecule 1 (VCAM-1); these molecules drive the routing of new macrophages from the periphery towards the endothelium and vascular sub-endothelium. Due to LDL captation, invasive macrophages of the subendothelial domain are transformed into foamy cells, an event which, together with the migration of muscle cells with a high ability to express TNF-α, IFN-γ, IL-1β, and IL-6, is key to the thickening of the atherosclerotic platelet and the increase of cardiovascular risk in the patient.23 Furthermore, not only have the oxLDLs contributed to atherosclerosis, but numerous lipids and oxidised phospholipids have been identified in atherosclerotic platelets, among which the platelet activating factor is of note (PAF), the oxidised phospholipids and lysophosphatidylcholine, which are themselves alone capable of inducing the expression of TNF-α, IL-1β, IL-6, TLR-2, TLR-4 and NF-κB in macrophages, and MCP-1 and VCAM in endothelial cells.23–25 Moreover, other molecules, such as pCr may be deposited in the intima layer of the artery and recruitment of monocytes-macrophages with inflammatory activity may take place through low affinity receptors for peripheral IgG.26 Several studies also suggest that IL-6 may play an important role as a lipidaemia regulator, since the blocking of this inflammatory mediator's function through the use of tocilizumab improves the relationship between total cholesterol and the high density lipoproteins (HDL).27 Total evidence therefore suggests that lipid disruption is currently considered a clear inducer of meta-inflammation in atherogenesis, because: (1) LDLs and oxLDLs are clear inflammatory stimulae, (2) atherosclerosis does not develop in animal models or in patients where without a significantly high level of these molecules, and (3) clearly inflammatory multiple molecules such as pCr and IL-6 may regulate the atherosclerotic process.27 In this apparent triad of dyslipidaemia – low-grade systemic inflammation – atherogenesis, one of the topics of greatest interest for its possible medical application is the prognostic value of anti-oxLDL antibodies in atherosclerosis, since patients with coronary disease present a reduction in anti-oxLDL anti-bodes leading to greater severity of acute ischaemic syndrome.28 Furthermore, the infusion of anti-oxLDL antibodies reduces atherogenesis in mice which are susceptible to the development of atheroesclerosis.29 However, a neutralisation test of these antibodies in prospective patient cohorts needs to be performed to determine their true prognostic value and reach a more in-depth understanding of the role of immune response in atherogenesis.
Diabetes mellitus type 2 and low-grade systemic inflammationDiabetes mellitus type 2 is a disease of multifactorial aetiology characterised by chronic hyperglycaemia associated with resistance to insulin activity and an insufficient compensatory response in the secretion of this hormone. A great deal of research studies indicate that the physiopathogenesis of this illness is closely related with the systemic inflammatory process, which could be active prior to the development of clinically detectable metabolic changes (Fig. 2).30 Even the children of people with diabetes mellitus type 2 present high inflammatory markers long before they present with any metabolic changes.31

Low-grade systemic inflammation contributes to the development of insulin resistance in several ways. AGE: advanced glycation end products; FFA: free fatty acids; AKT: protein kinase B; RNS: reactive nitrogen species; ROS: reactive oxygen species; GLUT: glucose carrier proteins; IL: interleukin; LGSI: low-grade systemic inflammation; IRS: insulin receptor substrate; NF-κβ: nuclear factor kappa beta; oxLDL: oxidised low density lipo proteins; RAGE: receptor advanced glycation end products; TLR: toll-like receptor; TNF: tumour necrosis factor.
In diabetes mellitus type 2 inflammation of the pancreatic islet plays an essential role. There are two pathways with regards to the inflammatory process in the pancreatic β: (1) activation of TLR-2 and TLR-4 by metabolic DAMP such as AGEs, and (2) NLRP3 inflammosome assembly.32 The result of this process is insulitis, characterised by the continuous release of IL-6, IL-8, TNF-α and MCP-1, the activation of insular macrophages and the recruitment of new peripheral monocytes-macrophages. Other molecules such as IL-12, IL-17 and NADPH oxidase-1 also have a major part to play in islet inflammation.32 In insulitis, the pancreative β cell is most susceptible to the effects of IL-1β, since it expresses higher levels of the receptor of this inflammatory cytokine (IL-1R1).33 As predicted by our group 2011,34 blockage of the effects of IL-1β with the monocolonal antibody Gevokizumab led to an improvement in the secretion of peptide C, thereby lowering levels of glycosylated haemoglobin and inflammatory cytokines in patients with diabetes mellitus type 2.35
This inflammatory process also simultaneously affects other tissues through a cascade of events which currently constitute one of the main physiopathogenic theories in the development of insulin resistance and diabetes mellitus type 2. During low-grade systemic inflammation, the adipose, hepatic and muscular tissue are exposed to infiltration of inflammatory macrophages which are the producers of TNF-α, a cytokine which directly interferes with the ability of these tissues to respond to insulin. Under normal conditions insulin adheres to a receptor located on the surface of the adipocyes, hepatocytes and miocytes, initiating an intracellular signalling cascade mediated by the insulin receptor substrate (IRS), which activates AKT and finally induces the mobilisation of glucose transporter proteins (GLUT-2 and 4) for the incorporation of this glucose to the inside of the cell. In the presence of TNF-α, activation of the IRS-AKT-GLUT signalling is inhibited by the action of the responsive intracellular pathways to TNF-α, including ERK/JNK, PTP1B and NF-κB, which in turn directly compete with or inhibit phosphorylation of IRS-2, AKT, GLUT-2 and 4.36 To sum up, this inflammation leads to a state of hyperglycaemia and later a compensatory state of hyperinsulinaemia, followed by claudication and apoptosis of the pancreative β cell, insulitis, reduction in insulin levels and the establishment of diabetes mellitus type 2. As previously mentioned, hyperglycaemia per se leads to a state of systemic inflammation characterised by the activation of macrophages which infiltrate the tissue and concomitantly raise inflammatory cytokine levels, which perpetuate low-grade systemic inflammation and its metabolic consequences.
Arterial hypertension as a result of multiple inflammatory mechanismsAs a clinical entity, systemic arterial hypertension presents between 20 and 50 years of age, with a prevalence of up to 75% in advanced ages.37 Systemic arterial hypertension is closely related to diseases such as: vascular disease of the brain, coronary disease, heart failure, auricular fibrilation, and kidney failure. Different molecular mechanisms converge in the development of the systemic arterial hypertension clinical phenotype where the inflammation appears to play a central role in the thickening of the intima and media layers and also arterial stiffness in addition to being highly important in endothelial dysfunction.
Changes in the expression profile of the vascular endothelium have been associated with higher adhesion and atherogenicity. These changes include the secretion of molecules such as soluble endothelial selectin, thrombomodulin, VCAM-1, ICAM-1 and the Von Willebrand factor,31,32 and appear as a consequence of the stimulus of several metabolic DAMPs such as FFAs, particularly unsaturated fatty acids. These molecules have a negative effect on endothelial function on activating NF-κB and IL-6 and facilitate the production of ROS.32 One of the most prevalent inflammatory molecules involved in the development of systemic arterial hypertension is pCr.38 In a study involving 320 non diabetic patients recently diagnosed with hypertension where higher pCr, soluble RAGE (sRAGE) and asymmetrical dimethylarginine levels were observed, it was noted that the highest sRAGE rates correlated with a higher body mass index, higher blood pressure and higher levels of pCr.39 The pCr levels reduced the production of ON due to its action on endothelial ON synthase (eNOS) and induced a greater expression of type 1 angiotensine receptors (ATR1) in vascular smooth muscle, facilitating activation of NF-κB by angiotensis II and the release of TNF-α, with negative impact on eNOS.40 It is of note that pCr also inhibits prostacyclin synthase, which interferes with the prostacyclin-mediated vasodilator effect.41 In aortic human endothelial cells, pCr inhibits GTP cyclohydrolase, lowering tetrahydrobiopterin synthesis (BH4, co-factor of eNOS) and stimulating NADPH oxidase, which increases ROS levels. The final effect is reduction in ON production and thereby in hypertension. In patients who lack morning blood pressure control, having an elevated serum pCr level increases cardiovascular risk by 5.77 times (IC 95% 2.11–15.81) in comparison with those who have a normal pCr level.42 The atheromatous plaques in patients without early morning blood pressure control, compared with those who do not lack control, are higher in inflammatory activity, characterised by a larger amount of macrophages and T infiltrating lymphocytes, as well as production of NF-κB and TNF-α. This inflammatory phenotype is associated with greater activity of ubiquitin–proteasome, the main non-lysosomal intracellular pathway of protein degradation marked for degradation by proteasome activity.43 This pathway is necessary for NF-κB activation as it degrades the NF-κB kinase inhibiting proteins (IKK). In hypertensive animal models it has been determined that the infiltration of tissues by macrophages and NF-κB and IL-1β production is more intense in the kidney, which stimulates renin–angiotensin–aldosterone system activation and plays a part in kidney injury induced by hypertension.44
Recognition of systemic arterial hypertension as a systemic inflammatory process is fundamental not only for understanding the genesis of the disease but also the development of any complications associated with it. From an immunological viewpoint treatment approach could therefore have patient benefits. For example, in patients with heart failure secondary to systemic arterial hypertension, the beneficial effects of carvedilol are lower in those who have higher levels of inflammatory cytokines such as TNF-α and IL-18.45 To sum up, low-grade systemic inflammation not only plays a part in the development of systemic arterial hypertension but is also associated with its severity and contributes to the development of complications such as hypertrophy and left ventricular dysfunction, arrhythmogenesis and atherosclerosis. Research is therefore essential for reaching a better understanding of the disease and an optimum approach for patient care.
ConclusionsThe evidence presented suggests that low-grade systemic inflammation plays an essential role in the development of metabolic diseases, such as dyslipidaemia, atherogenesis, diabetes mellitus type 2 and systemic arterial hypertension. This meta-inflammation causes activation of intracellular signals leading to the releases of local and systemic inflammatory factors, which feedback and interconnect pro-inflammatory producers of inflammatory cytokines and ROS macrophages to adipose, muscular and liver tissues. Once established, the low-grade systemic inflammation promotes and perpetuates metabolic changes establishing a vicious circle leading to pathological processes such as insulin resistance, atherosclerosis and endothelial dysfunction. Breaking the cycle therefore depends on simultaneously controlling both metabolic and inflammatory components. Future research must therefore aim at designing anti-inflammatory therapies leading to appropriate metabolic regulation, whilst taking into account cytokine and circulating immune cell levels, factors of transcription and adhesion, and the patient's metabolic DAMP profile. This more integrated approach, together with clinical data, could contribute to the identification of different meta-inflammation phenotypes depending on the level and relationship between their components, which, in turn, could be clinically identified by biomarkers for predicting a patient's evolution with a metabolic disease and establish customised anti-inflammatory therapies based on molecular evidence.
Conflict of interestsThe authors have no conflict of interests to declare.
We would like to thank the experimental biologist Angélica Fabiola Barragán for her critical review of this article and technical assistance in the design of the tables.
Please cite this article as: León-Pedroza JI, González-Tapia LA, del Olmo-Gil E, Castellanos-Rodríguez D, Escobedo G, González-Chávez A. Inflamación sistémica de grado bajo y su relación con el desarrollo de enfermedades metabólicas: de la evidencia molecular a la aplicación clínica. Cir Cir. 2015;83:543–551.