


El cáncer de tiroides es el tumor maligno más frecuente del sistema endocrino; la variante papilar representa entre el 80 y el 90% de todos los casos diagnosticados. En el desarrollo del cáncer papilar de tiroides están afectados principalmente 2genes llamados BRAF y RAS, que alteran el sistema de señalización intracelular de las proteínas conocidas como «cinasas de proteínas activadas por mitógenos» (MAPK, por sus siglas en inglés para mitogen-activated protein kinase) y que se componen de «módulos» de proteínas de señalización interna (Receptor/Ras/Raf/MEK/ERK), que van de la membrana celular al núcleo y que en el cáncer de tiroides regulan diversos procesos celulares, como la diferenciación, crecimiento, desarrollo y apoptosis. Tienen un papel importante en la patogénesis del cáncer de tiroides, debido a que son usados como biomarcadores moleculares, como elementos de diagnóstico, pronóstico y como posibles blancos terapéuticos moleculares.
ObjetivoRevisar los mecanismos moleculares que intervienen en las vías de señalización, en las que están involucradas las proteínas de los genes BRAF y RAS, en el cáncer de tiroides.
ConclusionesLas mutaciones en el gen BRAF han sido correlacionadas con una pobre respuesta al tratamiento con quimioterapia tradicional, además de ser un índice de mal pronóstico. La terapia molecular es de gran interés en este tipo de cáncer, ya que se han desarrollado medicamentos que actúan inhibiendo alguno de los componentes de la vía de señalización (RET/PTC)/Ras/Raf/MEK/ERK, con especial énfasis en la sección (RET/PTC)/Ras/Raf, que constituye un efector principal de la vía ERK.
Thyroid cancer is the most common malignancy of the endocrine system, the papillary variant accounts for 80-90% of all diagnosed cases. In the development of papillary thyroid cancer, BRAF and RAS genes are mainly affected, resulting in a modification of the system of intracellular signaling proteins known as «protein kinase mitogen-activated» (MAPK) which consist of «modules» of internal signaling proteins (Receptor/Ras/Raf/MEK/ERK) from the cell membrane to the nucleus. In thyroid cancer, these signanling proteins regulate diverse cellular processes such as differentiation, growth, development and apoptosis. MAPK play an important role in the pathogenesis of thyroid cancer as they are used as molecular biomarkers for diagnostic, prognostic and as possible therapeutic molecular targets. Mutations in BRAF gene have been correlated with poor response to treatment with traditional chemotherapy and as an indicator of poor prognosis.
ObjectiveTo review the molecular mechanisms involved in intracellular signaling of BRAF and RAS genes in thyroid cancer.
ConclusionsMolecular therapy research is in progress for this type of cancer as new molecules have been developed in order to inhibit any of the components of the signaling pathway (RET/PTC)/Ras/Raf/MEK/ERK; with special emphasis on the (RET/PTC)/Ras/Raf section, which is a major effector of ERK pathway.
El cáncer de tiroides es el tumor maligno más frecuente del sistema endocrino y representa cerca del 1% de los casos de cáncer diagnosticados a nivel mundial. El cáncer diferenciado de tiroides incluye el tipo papilar y el folicular. El más frecuente es el carcinoma papilar, el cual constituye cerca del 80-90% de todos los casos diagnosticados y se presenta con mayor frecuencia en mujeres que en hombres, con una relación de 2:1. Su incidencia a nivel mundial presenta un incremento importante en las últimas 3décadas, probablemente debido a que los sistemas de diagnóstico han avanzado considerablemente, lo que ha permitido establecer diagnósticos más oportunos, así como implementar tratamientos cada vez más precisos1–5. Reportes recientes indican que existe una prevalencia similar en las edades de diagnóstico, que son independientes del grupo étnico; sin embargo, existen diferencias en la prevalencia por sexo y etnia: se ha reportado que hombres y mujeres caucásicos presentan una prevalencia de 6.3 y 7.1%; la población inglesa reporta 4.3 y 8.4%; la hispana 4.2 y 6.7% y la asiática 3.4 y 6.4%, respectivamente6. Se piensa que factores genéticos, influencias del medio ambiente y el acceso a los servicios de salud pueden ser los factores que determinen la incidencia del cáncer de tiroides en una región particular. Sin embargo, en la última década se ha observado un incremento sostenido en la tasa de cáncer de tiroides a nivel mundial, principalmente el del tipo papilar6.
En el año 2008 se reportaron 3,195 casos de cáncer de tiroides (1,351 en varones y 1,844 en mujeres), en México, lo que representó el 2.5% del total de neoplasias malignas, con una incidencia de 3 por 100,000 habitantes y una mortalidad de 0.6 por 100,000 habitantes, ubicándose como la sexta causa de muerte en mujeres y la decimotercera en hombres, con frecuencia máxima entre los 41 y los 50 años: el 60% de los casos acontecen entre los 31 y los 60 años. De las enfermedades tiroideas, el carcinoma papilar y sus variantes representan el 80.3% y el cáncer folicular y sus variantes, el 2.4%7–9.
A nivel molecular, el cáncer papilar de tiroides frecuentemente se presenta con alteraciones metabólicas en los sistemas de señalización intracelular, que involucran la activación de proteínas conocidas como cinasas de proteínas activadas por mitógenos (MAPK, por sus siglas en inglés para mitogen-activated protein kinase) que, en los últimos años, han sido motivo de muchos estudios por su papel en la patogénesis del cáncer de tiroides y su uso como biomarcadores moleculares, además de su potencial uso como elementos de diagnóstico, pronóstico y su posible uso como blancos terapéuticos moleculares10–12.
Alteraciones genéticas en el cáncerSe sabe que alteraciones en 3tipos de genes pueden ser responsables del inicio y mantenimiento del cáncer: a) oncogenes, b) genes supresores tumorales y c) genes de estabilidad. También se sabe que la célula posee diversos mecanismos para salvaguardar y proteger al organismo de un efecto letal, potencialmente cancerígeno, como puede ser una mutación genética y, por eso, solo cuando existen alteraciones en varios genes es cuando se desarrolla cáncer12–14.
Una mutación en un oncogén da como resultado que un gen constitutivo que debería permanecer activo o inactivo en forma normal, se inactive o active. La activación de un oncogén puede ser por: translocación cromosomal, amplificación anómala de un gen o por mutación intragénica, que afecta un residuo crucial para la actividad de dicho gen. Una mutación en un alelo de un oncogén generalmente es suficiente para conferir un descontrol en el crecimiento celular. Por su parte, los genes supresores tumorales actúan en forma opuesta a lo que puede ser una mutación genética: en su caso, reducen la actividad del gen y su producto. Tal inactivación surge como producto de mutaciones que hacen perder residuos esenciales para la actividad de la proteína codificada por dicho gen, lo que a su vez resulta en proteínas truncadas ya sea por deleción, inserción o por alteraciones epigenéticas. La tercera clase de genes cancerígenos son llamados genes de estabilidad o «vigilantes». En su caso, este tipo de genes promueven la tumorogénesis por caminos distintos de los otros 2tipos de genes tumorales mencionados. En este tipo de genes «vigilantes», se encuentran aquellos que se encargan principalmente de reparar todas aquellas alteraciones que resultan de la replicación del ADN, cuando esta ocurre de manera normal o es expuesto a un mutágeno. Además, otros genes de estabilidad controlan los procesos que involucran grandes porciones de cromosomas, tales como los que regulan la recombinación y segregación cromosomales que ocurren durante la mitosis. Los genes estabilizadores mantienen al mínimo las alteraciones genéticas y, cuando son inactivados, existe un aumento en la tasa de mutaciones14–17.
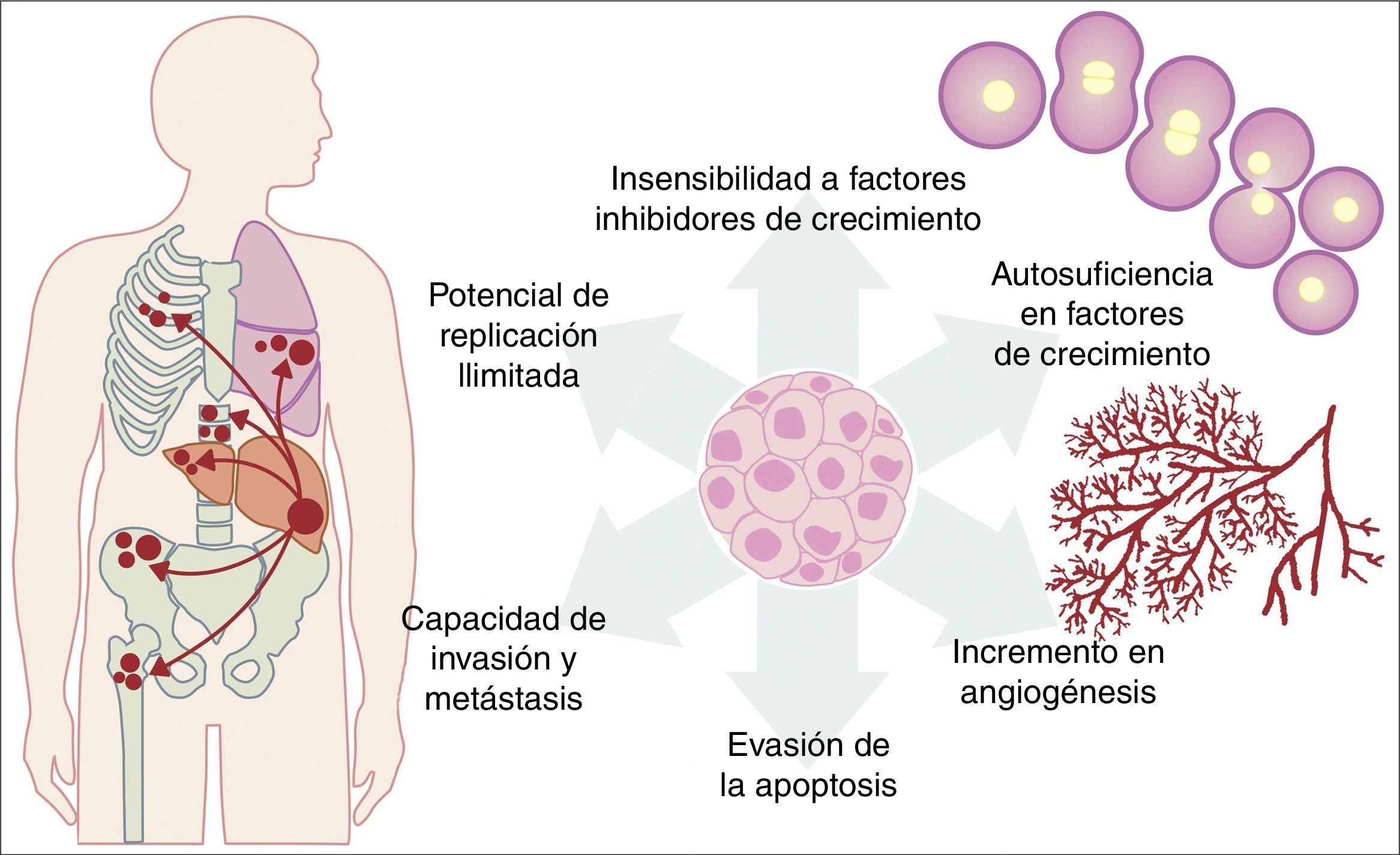
Todas las mutaciones anteriormente descritas funcionan igual que un proceso fisiológico celular normal. El crecimiento celular cancerígeno puede ser provocado por activación de oncogenes, que controlan el ciclo celular al inhibir a la muerte celular (apoptosis), o incrementando la transición de células del estado G0 a G1 («liberación de arresto celular»), o facilitando la provisión de nutrientes al aumentar fenómenos como la angiogénesis. Es importante recordar que una mutación se define como cualquier cambio en la secuencia del genoma y que, en estos cambios, se incluyen los que afectan a un par de bases, así como los que son pequeños o grandes cambios como son: deleciones, inserciones, amplificaciones o translocaciones. En las células cancerígenas se pueden presentar todos los tipos de mutaciones; sin embargo, se considera como enfermedad cuando la ocurrencia de una mutación posterior al nacimiento es patogénica14–17. Por otro lado, una vez que la célula se ha hecho cancerígena, presenta una serie de cambios metabólicos, moleculares y estructurales que le permiten sobrevivir, desarrollarse e, incluso, producir metástasis. Dentro de estos cambios generales encontramos una autosuficiencia en factores de crecimiento, una insensibilidad a los factores inhibidores del crecimiento, una capacidad de invasión a tejidos vecinos y de metástasis, un potencial de replicación ilimitada, un incremento en la angiogénesis y el desarrollo de mecanismos de evasión de la apoptosis (fig. 1)18. De manera importante, la hipótesis de Knudson ha contribuido a la comprensión de la forma en la que mutaciones en genes involucrados en el desarrollo de cáncer, las cuales tienen una forma de herencia autosómica dominante, que se deben a la pérdida de función de un gen supresor de tumor, generando la subsecuente mutación tejido-específica. Sin embargo, estas contribuciones fueron previas al creciente conocimiento que ahora se tiene sobre la regulación de los micro-ARN y ARN no codificantes, lo cual ha dado lugar al «modelo continuo» de regulación de genes supresores de tumores, en el cual no solo las mutaciones genéticas heredadas y las somáticas son las que contribuyen al desarrollo del cáncer, sino también los elementos epigenéticos, como lo son los micro-ARN, que contribuyen en el desarrollo del cáncer de manera significativa19.

Esquema que ilustra algunas de las características patológicas adquiridas por una célula cancerígena que le ayudan a sobrevivir en el organismo.
Fuente: Adaptado de Hanahan y Weinberg18.
En el cáncer de tiroides existen un número importante de alteraciones genéticas que se relacionan con la progresión y la diferenciación celular. Estas alteraciones pueden englobarse en 2categorías principales: 1) rearreglo cromosomal y 2) mutación puntual. Ambas alteraciones afectan principalmente a 2genes llamados BRAF y RAS. Las mutaciones de alguno de estos 2genes se han encontrado en más del 80% de los casos con carcinoma papilar de tiroides y raramente se encuentran traslapados en el mismo tumor. Estas mutaciones alteran el sistema de transducción de la señal intracelular, que regula diversas vías metabólicas, que repercuten directamente en procesos celulares como son la diferenciación, expansión y apoptosis, entre otras. De acuerdo con los datos actuales, las mutaciones que afectan a BRAF existen exclusivamente en el cáncer de tiroides papilar y en el cáncer de tiroides anaplásico, pero no han sido descritas para otros tipos histológicos de cáncer de tiroides, como son el cáncer folicular de tiroides y el cáncer medular de tiroides. Por otro lado, se ha descrito que la mutación BRAFV600E (sustitución de una valina por un ácido glutámico en la posición 600), se encuentra en cerca del 60% en todos los tipos de cáncer de tiroides. Las mutaciones producen una activación en las vías de señalización dependientes de MAP-cinasas que, se piensa, son un evento temprano en el desarrollo y progresión del cáncer de tiroides20,21.
La «cascada» de procesos de señalización intracelulares dependientes de MAP-cinasas son muy importantes, ya que regulan el número y participación de genes implicados en la proliferación, diferenciación y sobrevivencia de las células cancerosas. El sistema de MAP-cinasas se compone de «módulos» de proteínas de señalización, que van desde la membrana hasta el núcleo celular y que se conservan desde las levaduras hasta los vertebrados22–24. Este sistema es regulado a través de una «cascada» de fosforilaciones, en donde intervienen otras fosforilasas, localizadas en 2lugares en relación con la MAP-cinasa: 1) «corriente-arriba» y 2) «corriente-abajo»20–25.
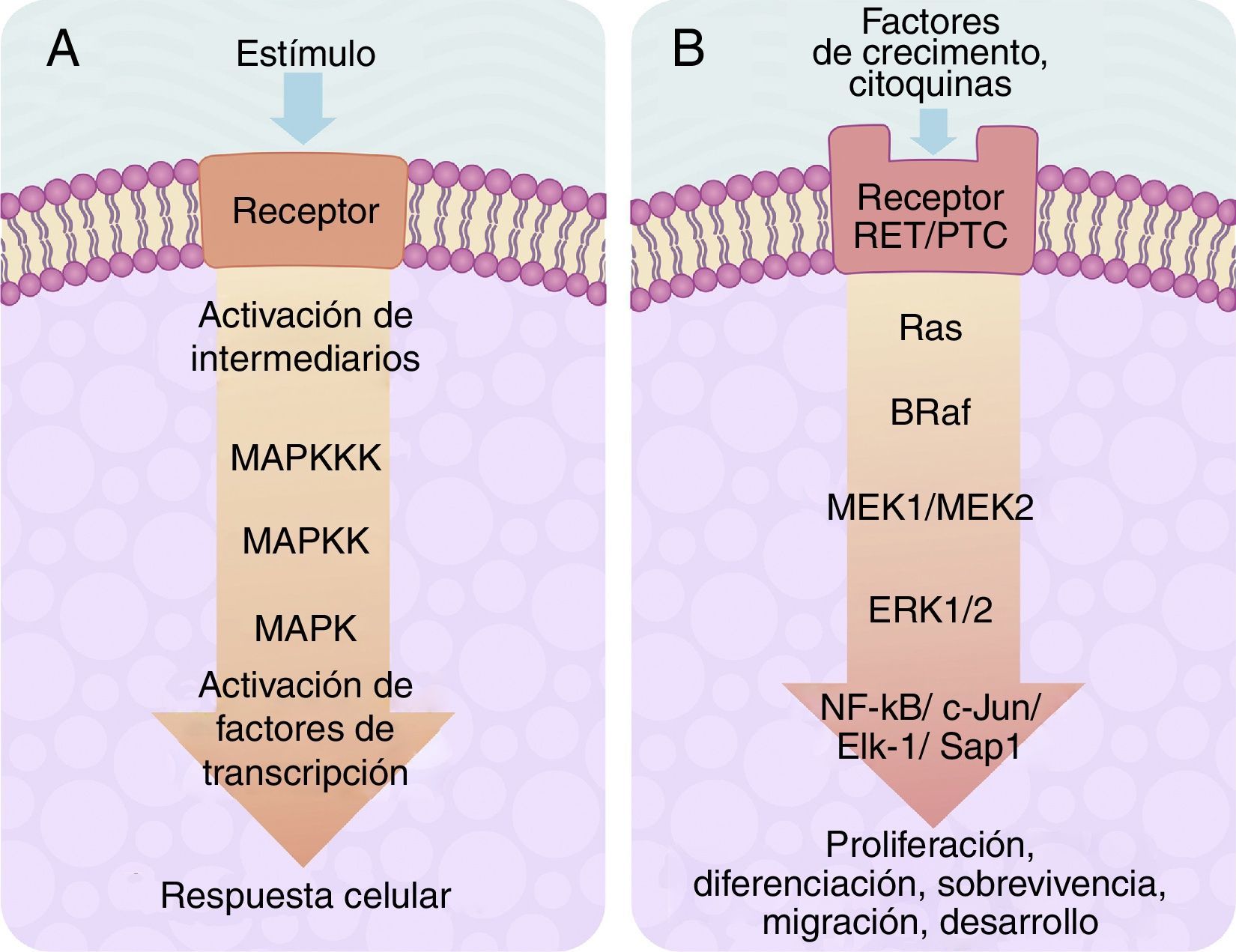
El centro de activación de cualquier señal metabólica encadenada en «cascada» para activar a una MAP-cinasa se compone, «corriente arriba», de 3niveles de cinasas, que se activan en secuencia a través de la señal de activación de un receptor de membrana con actividad de tirosincinasa (fig. 2A). En células de eucariontes, existen múltiples vías de señalización dependientes de MAP-cinasas, que son activadas por diversos mecanismos de estímulos. En la actualidad, se han definido al menos 4diferentes tipos de subfamilias de MAP-cinasas, que son: ERK-1/2, JNK-1/2/3, P38-a/b/g/d y ERK-5. De gran importancia en medicina y, sobre todo, para la mejor comprensión de los mecanismos moleculares de generación y regulación del cáncer de tiroides, se encuentra la vía de señalización ERK-1/2 MAP-cinasa, ya que es preferentemente activada como respuesta a un estímulo mitogénico, tal como la activación de un receptor para un factor de crecimiento (fig. 2B). La vía de señalización ERK-1/2 MAP-cinasa se activa inicialmente «corriente arriba» con la activación de un receptor de membrana, al que se le ha unido su ligando. El receptor ahora activa a una proteína de tipo G llamada Ras (que tiene actividad de cinasa), la cual «recluta» mediante fosforilaciones subsecuentes a una familia de MAPKKK llamadas Raf que, a su vez, activan a una serie de MAPKK llamadas MEK-1/2. Finalmente, MEK-1/2 activan a ERK-1/2 el cual es translocado dentro del núcleo, en donde fosforila a factores de transcripción de varios genes, llevando a cabo la regulación de su expresión. Esta translocación nuclear es requerida en respuesta a estímulos como son los factores de crecimiento, que regulan la entrada al ciclo celular o la diferenciación celular. De ahí la importancia de su estudio para fenómenos como el cáncer, en donde existe un descontrol de los procesos celulares ya mencionados26–29.
 Esquema de una cascada de señalización intracelular activada por receptor de membrana el cual activa «corriente abajo» una serie de cinasas agrupadas una tras otra en secuencia MAPKKK-MAPKK-MAPK y termina en la activación de un factor de transcripción que origina una respuesta celular. B) Esquema de activación del factor de transcripción ERK-1/2 a través del sistema de señalización intracelular de cinasas en el cáncer de tiroides. La activación inicial es a través de un receptor de membrana tipo tirosincinasa (RET/PTC) y se continúa «corriente abajo» a través de la secuencia de las cinasas Ras/B-Raf/MEK-1/2.")
A) Esquema de una cascada de señalización intracelular activada por receptor de membrana el cual activa «corriente abajo» una serie de cinasas agrupadas una tras otra en secuencia MAPKKK-MAPKK-MAPK y termina en la activación de un factor de transcripción que origina una respuesta celular. B) Esquema de activación del factor de transcripción ERK-1/2 a través del sistema de señalización intracelular de cinasas en el cáncer de tiroides. La activación inicial es a través de un receptor de membrana tipo tirosincinasa (RET/PTC) y se continúa «corriente abajo» a través de la secuencia de las cinasas Ras/B-Raf/MEK-1/2.
Se ha considerado que el proceso de tumorogénesis requiere de la presencia de una desregulación de los procesos de señalización intracelular, que involucran a distintos niveles la cascada de las MAP-cinasas, en donde las células cancerosas adquieren diversas capacidades como son: 1) independizarse de las señales de proliferación; 2) evadir la apoptosis; 3) insensibilizarse a las señales antiproliferación; 4) adquirir un potencial ilimitado de replicación; 5) invadir y producir metástasis a otros tejidos y 6) producir angiogénesis, que les proporciona nutrientes y soporte vital30,18.
Se ha reportado que diferentes neoplasias muestran una alteración en la vía de señalización intracelular, constituida por la cascada de activaciones en secuencia de las cinasas Ras/Raf/MEK/ERK, en las que el efector último de la cascada (que es la proteína ERK-cinasa) es activado «corriente arriba» por alteraciones génicas (mutaciones) que pueden afectar a las distintas cinasas localizadas en alguno de los niveles de la cadena de reacciones, empezando por una sobreexpresión del receptor tipo cinasa de tirosina y, lo más frecuente, con mutaciones en las proteínas Ras y Raf. La cascada de señalización intracelular en una célula cancerígena empieza con la activación de un receptor de membrana (con actividad de cinasa de tirosina) cuando este se une a su ligando o por causa de un estímulo externo (p. ej. alteración de la ósmosis, estrés oxidativo, carencia de nutrientes, etc.). Una vez que el receptor ha sido activado, este «recluta», mediante fosforilaciones específicas, al primer efector de la cascada de señalización que es una MAPKKK que puede ser A-Raf, B-Raf o Raf-1. Estas cinasas fosforilan a las siguientes MAPKK que son MEK-1/2 y, finalmente, estas cinasas fosforilan a una MAPK, que es ERK-1 o ERK-2, las cuales son translocadas al núcleo para iniciar la activación/inhibición de genes específicos. Si bien esta cascada de señalización se encuentra de forma constitutiva en todas las células normales del organismo, en la célula cancerosa cada componente de la cadena puede presentar mutaciones que favorecen la sobreactivación de su cinasa «corriente abajo», lo que incrementa exponencialmente la actividad celular a través de la activación/inhibición de genes específicos. Así, por ejemplo, ha sido descrito que las mutaciones en las cinasas Ras y Raf (que son los componentes de la vía intracelular compuesta por Receptor/Ras/Raf/MEK/ERK), se encuentran en todos los tipos de cáncer de tiroides, principalmente en el de tipo papilar, lo que indica que una única alteración en dichas moléculas puede ser suficiente para una transformación maligna de las células del tiroides.
De esta forma, se sabe que la presencia de la mutación llamada BRAFV600E (consistente en la presencia de un residuo de valina, en lugar de un residuo de ácido glutámico, en la posición 600 de la cinasa de proteína B-Raf), cuya prevalencia ha sido reportada entre el 27 y el 80% en distintas poblaciones de pacientes con cáncer de tiroides, resulta en un incremento de más de 400 veces la actividad de B-Raf, lo que produce un incremento en la actividad «corriente abajo» de los efectores ERK, que aumentan su capacidad fosforilante y, con ello, la actividad de diversos genes31 claves para la el metabolismo, proliferación y apoptosis celular.
Las mutaciones reportadas como más frecuentes en el cáncer papilar de tiroides son: rearreglos en el gen RET/PTC (receptor de membrana con actividad de tirosincinasa) y mutaciones en B-Raf y en Ras (proteínas con actividad de cinasa). Todos ellos están involucrados en la vía de señalización intracelular que activa al efector nuclear ERK. Estas alteraciones se encuentran exclusivamente en pacientes con cáncer papilar de tiroides, lo que muestra que cada alteración por separado puede ser suficiente para una transformación maligna de células tiroideas. El protooncogén llamado RET (de las siglas en inglés para rearranged during transfection), codifica para un receptor de membrana con actividad de cinasa de tirosina, el cual se expresa de manera particular en las células parafoliculares tipo C en el cáncer de tiroides, que deriva en proteínas aberrantes con diferentes formas quiméricas del receptor; sin embargo, su expresión es muy baja en las células foliculares. A estas formas quiméricas del receptor se les ha dado el nombre de RET/PTC (por las siglas del inglés para rearranged during transfection/papillar tyroid cancer associate). En la actualidad, han sido descritas más de 11tipos diferentes de proteínas RET/PTC, en donde RET/PTC1 y RET/PTC3 son las más frecuentemente encontradas en el cáncer de tiroides32,33.
En la célula cancerosa, RET/PTC activa «corriente abajo» en forma constitutiva a una cinasa llamada Ras. En la actualidad, se sabe que Ras activa una gran cantidad de moléculas que actúan como sistemas de señalización intracelular «corriente abajo» e inducen las propiedades de invasión de células cancerosas. Se ha reportado que Ras también activa las proteínas de reacción a estrés, como la cinasa Raf, la cual es el principal efector citosólico «corriente abajo» de Ras.
Se han reportado varias isoformas de Raf: A-Raf, B-Raf y Raf-1 (también llamada C-Raf). La activación de Raf incluye una serie de pasos metabólicos altamente regulados, que inician con su reclutamiento a la membrana interna celular por la proteína Ras. Una vez que Raf se «activa», pueda asociarse «corriente abajo» con otras cinasas, entre ellas MEK-1 y MEK-2. Para su activación, además de que Ras esté activado, las isoformas de Raf requieren de una enzima con actividad de fosfatasa de proteínas conocida como Src-cinasa. Esta proteína fosforila o desfosforila determinados residuos de aminoácidos, localizados dentro de los dominios de Raf, que regulan su estado de inactivo/activo. De las 3isoformas conocidas para Raf, la isoforma B-Raf es la que presenta una actividad basal más elevada en comparación con A-Raf o Raf-1. Lo anterior es muy importante, ya que en el cáncer de tiroides se han descrito una gran cantidad de mutaciones en la proteína B-Raf, en comparación con A-Raf o Raf-1, lo que permite usar a B-Raf como un marcador tumoral para diversos tipos de cáncer, particularmente de tiroides. Por otro lado, B-Raf es el más potente activador «corriente abajo» de la cinasa MEK-1, la cual es el efector de ERK.
La proteína MEK-1 es una cinasa de proteína con actividad dual, puesto que puede fosforilar/desfosforilar residuos de serina y tirosina. Una vez que MEK-1 ha sido activada por B-Raf, se produce la activación «corriente abajo» del efector ERK, el cual es una cinasa de serina/treonina que fosforila diversas proteínas, tanto citosólicas como nucleares. De esta forma, la hiperactivación de la cascada de señalización que regula a ERK puede inducir el arresto del ciclo celular. Por otro lado, si existe una aberración en cualquiera de las secuencias de la cascada de señalización, se puede inducir una trasformación tumoral de la célula. Así, la cinética de respuesta y su amplitud ante diversos ligandos o estímulos extracelulares pueden hacer que ERK regule de manera específica diversos programas biológicos como son la diferenciación y proliferación celular, o la apoptosis33.
Vía de señalización intracelular PI3K/Akt/mTOR en el cáncer de tiroidesAdemás de la vía de señalización intracelular ya señalada con anterioridad, también existe la vía fosfatidilinositol-3 cinasa/proteín cinasa B (Atk)/blanco de mamífero, para rapamicina (mTOR), también conocida como PI3K/Akt/mTOR. Dicha vía de señalización está involucrada en procesos como: la diferenciación y crecimiento celular, progresión del ciclo celular, endocitosis, motilidad, apoptosis y el metabolismo intermedio (relacionado principalmente con la captación de glucosa)34–36. En los últimos años, se han descrito alteraciones en dicha vía en diversos tipos de cáncer como el de tiroides, el gástrico, el neuroendocrino y el de ovario. Por otro lado, se ha observado que la activación de esta vía en células cancerígenas puede incrementar la resistencia al tratamiento con cis-platino, cabo-platino y paclitaxel, por lo que su estudio puede ayudar a entender los mecanismos de génesis y progresión del cáncer, así como los mecanismos de resistencia tumoral para ser un posible blanco, para la terapia molecular y el manejo del cáncer de tiroides34–38.
La fosfatidilinositol-3 cinasa (PI3K) fosforila a grupos 3-OH del anillo inositol de grupos fosfatidilinositoles. Los subproductos de la reacción de la PI3K medían reversiblemente la localización membrana-citoplasma de proteínas, que contienen dominios de unión a lípidos complejos y un incremento en su actividad por arriba de lo normal, asociada con una transformación celular oncogénica37,38.
En términos de señalización intracelular, la PI3K clase I puede ser un efector «corriente abajo» ya sea de un receptor de membrana con actividad de tirosincinasa o de una proteína G unida a un receptor38,39. Por otro lado, la proteína conocida como Akt tiene un peso molecular aproximado de 57kDa, que es una cinasa de serina/treonina que pertenece a la familia de cinasas en donde se encuentran: la proteincinasa A, proteincinasa G y proteincinasa C. La Akt también es conocida como proteincinasa B (PKB)38–42.
La proteína llamada mTOR es una serina/treonina cinasa codificada por el gen FRAP 1 y es el principal efector «corriente abajo» del complejo PI3K/Akt. Existen 2distintas isoformas de mTOR, llamadas mTORC1 y mTORC2. Cada una es un multicomplejo proteico formado por varios componentes, en donde el centro es la proteína mTOR en sí misma, a la que se le agregan varias subunidades. El complejo mTORC1 está formado por la proteína mTOR asociada con la proteína RAPTOR, que funciona como andamio y regula positivamente a mTOR. También está asociado a 2reguladores negativos que son PRAS40 y la proteína DEPTOR, mientras que la subunidad mLST8, unida al complejo mTORC1, regula su actividad40–43. La vía de comunicación intracelular mediada por mTORC1 está involucrada en el crecimiento y diferenciación celular, mientras que el complejo mTORC2 confiere insensibilidad a la rapamicina42,43. La desregulación de la actividad de mTOR está correlacionada con la presencia de hamartomas, esclerosis tuberosa, síndrome de Peutz-Jeghers, así como con diversos tipos de cáncer en los que las alteraciones genéticas a los genes reguladores de mTOR, tales como TSC1-TSC2 y LKB1, presentan diversos tipos de mutaciones43–45.
Por otro lado, durante la formación del hueso, el gen RUNX2 es determinante para la diferenciación del osteoblastoma, así como la proliferación de los condrocitos y la diferenciación e hipertrofia endocondral. Se ha reportado su participación como promotor de tumores en el cáncer de mama y próstata, además de asociarse con genes que controlan la invasión y metástasis tumoral, ya que promueve la expresión de las metaloproteinasas MMP2, MMP13, MMP14. De esta forma, RUNX2 se ha definido como un factor de transcripción promigratorio, proinvasivo y proangiogénico, además de participar en los eventos iniciales de la tumorogénesis y dirigir la invasión de las metástasis óseas44–46.
La expresión y la actividad de RUNX2 es inducida por Akt, en forma directa e indirecta. Experimentos hechos in vitro con células cancerosas han demostrado que la activación de la vía de comunicación intracelular PI3K/Akt produce directamente un incremento de la afinidad al ADN por los factores RUNX2 y RUNX2-dependiente de transcripción y que las mutaciones que afectan a Akt producen una disminución en la afinidad de RUNX2, lo que afecta la progresión del ciclo celular. En forma indirecta, la activación de la vía PI3K/Akt regula la actividad de RUNX2, ya sea incrementando su estabilidad proteica o actuando a través de un factor de transcripción nuclear llamado FoxO, un activador o represor transcripcional de genes específicos entre el núcleo y el citoplasma47,48.
Mutaciones y cáncer de tiroidesUna mutación en cualquier proteína involucrada en la cascada de señalización intracelular mediada por un receptor del tipo cinasa de tirosina puede determinar que una célula normal se convierta en célula cancerígena. En ese contexto, se ha descrito que cerca del 70% de los pacientes que presentan cáncer papilar de tiroides tienen al menos una mutación que afecta la cascada de activación de ERK; las más frecuentes son las que afectan a Ras y B-Raf y, en menor medida, a RET/PTC.
La prevalencia de mutaciones RET/PTC en el cáncer de tiroides es variada, ya que se ha visto que depende de la región geográfica, pero se puede decir que alrededor del 20% de los casos de cáncer de tiroides presentan estas alteraciones, así como que es más frecuente en personas jóvenes o con antecedentes de haberse sometido a tratamiento con radiación. Por otro lado, las mutaciones en algún miembro de la familia de genes que codifican para la proteína Ras han sido descritas principalmente en el adenoma folicular y el carcinoma de tiroides, pero únicamente han sido descritas en cerca del 10% de los casos de cáncer papilar de tiroides34.
En el caso de B-Raf (que también puede ser escrito como BRaf) se ha observado que se encuentra expresado de manera normal en: células hematopoyéticas, neuronas, células testiculares y en las células foliculares del tiroides. Al contrario de lo que pasa con las mutaciones en A-Raf y Raf-1 que son extremadamente raras, las mutaciones en B-Raf son las más frecuentes en el cáncer papilar de tiroides, además de que es la segunda mutación somática encontrada en todos los tipos de cáncer que afectan al ser humano. Así, por ejemplo, más del 45% de todos los tipos de cáncer que afectan al humano presentan una mutación en B-Raf. Cerca del 90% de esas mutaciones consisten en una sustitución de una timina por una adenina en el exon 15 del nucleótido 1799 (c.1799T>A), lo que da como resultado una mutación de B-Raf, en donde un residuo de valina es sustituido por un residuo de ácido glutámico en la posición 600 de la proteína. Dicha mutación es conocida como BRAFV600E (también escrita como B-Raf-V600E o BRAF-V600E). Se ha descrito que la prevalencia de esta mutación en el cáncer papilar de tiroides puede variar del 29 al 83%, dependiendo de la población estudiada, y no ha sido descrita en el cáncer folicular de tiroides49. Estudios realizados in vitro han demostrado que la sustitución de la valina por un ácido glutámico en la posición 600 adyacente a un residuo de treonina en la posición 599 dentro de la proteína B-Raf produce una conducta «similar» a una fosforilación, lo que hace que la estructura terciaria de la proteína se vea afectada en donde las interacciones hidrofóbicas se rompen entre el segmento P (el lugar de activación de la proteína por medio de fosforilación) y el segmento de actividad de cinasa, dando como resultado que la actividad de cinasa de BRAFV600E sea 460 veces más alta que B-Raf nativa. Esta elevada actividad constitutiva de BRAFV600E hace que se activen «corriente abajo» todos los efectores de la cascada de señalización hasta ERK, lo que resulta en la transformación de células normales en células cancerígenas, además de mantener su proliferación sin necesidad de la proteína Ras, para la activación de la vía de señalización.
Se ha demostrado que la presencia de BRAFV600E está relacionada con genes involucrados en el metabolismo del yodo, sobre todo los que tienen que ver con la captación de yodo por las células, para la síntesis de hormonas tiroideas. La presencia de BRAFV600E hace que las células tiroideas presenten alteraciones en varios de los genes involucrados en este proceso, los cuales han sido asociados a una mayor agresividad del tumor. De esta forma, la presencia de mutaciones en el gen BRAF ha sido correlacionada con una pobre respuesta al tratamiento con quimioterapia tradicional, además de ser un índice de mal pronóstico, ya que la mutación en BRAF ha sido encontrada tanto en células anaplásicas como en carcinomas pobremente diferenciados, lo que significa que los cambios moleculares en B-Raf se producen tempranamente en el fenómeno de tumorogénesis49.
Rearreglos en PAX8-PPARγ en cáncer de tiroidesEl gen PAX8 es un factor de trascripción específico de tiroides que regula su desarrollo y diferenciación. Por otro lado, el gen PPARγ actúa en el control del ciclo celular, la apoptosis y la carcinogénesis50, además del papel que desempeña en la adipogénesis y la sensibilidad a la insulina.
En conjunto, PAX8 y PPARγ tienen un papel muy importante en el cáncer de tiroides. La traslocación entre estos 2genes produce un nuevo gen, cuya sobreexpresión de la proteína de fusión resultante PAX8/PPARγ o PPFP altera la función de PPARγ de inhibidor «corriente abajo» de la proliferación celular e inductor de apoptosis. La translocación cromosomal PAX8-PPARγ ha sido detectada en el 35% del cáncer folicular de tiroides y en el carcinoma anaplásico de tiroides50. Además, en el cáncer folicular de tiroides la sobreexpresión de PAX8/PPARγ se asocia a la activación de MAP-cinasas dependientes de los receptores de TGFß, lo cual está íntimamente relacionado con el proceso de tumorogénesis50,51.
Terapia molecular en el cáncer de tiroidesUn carcinoma de tiroides, ya sea papilar o folicular, que esté bien diferenciado, tiene un comportamiento clínico bastante prometedor ya que puede ser tratado con cirugía, seguida de radioterapia. Sin embargo, los tumores que son indiferenciados o aquellos que pierden su capacidad de captar yodo radiactivo no pueden retirarse quirúrgicamente, por lo que son tumores con un pronóstico poco favorable. Este tipo de tumores son candidatos a ser tratados mediante terapia molecular, en donde la cadena de señalización intracelular (RET/PTC)/Ras/Raf/MEK/ERK es un blanco frecuente para estudios de terapia molecular.
La terapia molecular es de gran interés en este tipo de cáncer, ya que se han desarrollado medicamentos que actúan inhibiendo alguno de los componentes de la vía de señalización (RET/PTC)/Ras/Raf/MEK/ERK; con especial énfasis en la sección (RET/PTC)/Ras/Raf, ya que dicho segmento de la cascada de señalización es el principal efector de ERK. De esta forma, se ha descrito un compuesto como el ZD6474 que es un inhibidor de la actividad de la cinasa de RET/PTC, que ha sido eficaz en estudios in vitro y preclínicos para inducir arresto del ciclo celular en células de carcinoma papilar humano, lo que impide su crecimiento cuando son inyectadas en ratones52. También han sido probados en estudios in vitro, in vivo y preclínicos los compuestos pirazolopiridimidina (PP1 y PP2) y el sunitinib (SU12248), que nulifican la señal de RET/PTC para eliminar el efecto tumorogénico en animales de experimentación. También se han reportado resultados prometedores en estudios clínicos de fase ii para tratar pacientes que son refractarios al tratamiento con yodo radiactivo o con tumores irresecables quirúrgicamente, así como en los que presentan cáncer medular de tiroides53–55.
Por otro lado, se ha descrito que los compuestos GDC-0879 y PLX4720 inhiben selectivamente in vitro la actividad de cinasa de BRAFV600E, lo que hace que las células tumorales disminuyan su proliferación. Sin embargo, solo han probado ser efectivos en estudios preclínicos. Por otro lado, se ha observado que los pacientes que presentan la mutación BRAFV600E pueden llegar a desarrollar resistencia a este tipo de compuestos, ya que incrementan la expresión de Raf-1 por medio de la activación de Ras (que generalmente está sobreexpresada), con lo que la cascada de señalización «corriente abajo» hacia ERK puede seguir activa. Este hecho pone de manifiesto la necesidad de una genotipificación exacta para cada tipo de tumor, ya que es necesario asegurarse de que han sido detectadas las posibles mutaciones en alguno o varios de los componentes de la cadena de señalización que activan a ERK, puesto que, aunque un componente sea nulificado, otro puede ser activado y continuar con el estímulo de proliferación tumoral. Lo anterior también pone de manifiesto que el tratamiento debe darse con varios compuestos a la vez, para asegurar un mejor resultado inhibitorio de la cascada de señalización, máxime cuando se ha demostrado una resistencia de los tumores a la quimioterapia única que, en el caso de que presenten la mutación BRAFV600E, este tipo de tratamiento es el más recomendado54,55.
Por otro lado, la vía de señalización de PI3K/Akt/mTOR constituye otro blanco molecular atractivo, debido al beneficio terapéutico reportado para diferentes tumores malignos. La vía de PI3K/Akt/mTOR regula procesos celulares críticos como la proliferación, la apoptosis, el ciclo celular, el metabolismo y la angiogénesis, mientras que varios estudios preclínicos y clínicos apoyan que la inhibición farmacológica de PI3K/Akt/mTOR representa una estrategia bien tolerada y exitosa en el tratamiento de tumores malignos, como el cáncer de próstata, mama, colon, ovario, pulmón y melanoma56. Aparentemente, la disminución de la actividad de PI3K/Akt/mTOR se relaciona con la inducción de efectos de radiosensibilidad tumoral57, efectos proapoptóticos en células tumorales relacionados con una menor autofagia, así como la pérdida de troncalidad en células cancerígenas58. Respecto al cáncer de tiroides de tipo folicular, anaplásico avanzado o poco diferenciado, resistentes al yodo radiactivo y, por lo tanto, con mal pronóstico, por la respuesta al tratamiento convencional, varios estudios a nivel preclínico y clínico señalan que la inhibición de vía de PI3K/Akt/mTOR representa un blanco estratégico promisorio, ya sea como monoterapia o en terapia combinada, en estos tipos de cáncer de tiroides avanzados59.
ConclusionesEn los últimos 30 años, se han realizado importantes progresos en el campo de la biología molecular para tratar de discernir los mecanismos que están involucrados en el proceso de transformación tumoral, principalmente los estudios encaminados a entender los sistemas de señalización intracelular, en los que interviene la cascada de cinasas Ras/Raf/MEK/ERK. Esta cascada de señalización controla la proliferación celular en células normales y en células cancerosas. Además, son importantes vías de desarrollo de resistencia a los fármacos usados para quimioterapia.
Los estudios actuales han puesto de manifiesto que la cascada de señalización Ras/Raf/MEK/ERK puede promover la proliferación maligna, ya que estimula el crecimiento celular y, al mismo tiempo, inhibe la apoptosis (muerte celular programada), que es el mecanismo de control natural de la proliferación celular. Por otro lado, el descubrimiento de que las mutaciones en el gen BRAF pueden por sí solas promover la transformación y proliferación celular en diversos tipos de cáncer, principalmente el de tiroides, han puesto de manifiesto la necesidad de realizar más estudios sobre los mecanismos de control de este tipo de genes y de sus productos, lo que conlleva el desarrollo de agentes químicos eficaces para nulificar a este tipo de proteínas y, de esta forma, controlar el crecimiento y desarrollo de las células cancerígenas60. Lo anterior también pone en evidencia la necesidad de un mayor estudio de la genotipificación de los tumores, por su importancia en el uso de terapia molecular. La descripción de las mutaciones en BRAF puede predecir la sensibilidad de B-Raf a diversos agentes anticancerosos e inhibir «corriente abajo» a ERK y, de esta forma, inhibir el crecimiento y el desarrollo tumoral. La genotipificación del tumor también es útil para tratar de predecir el comportamiento clínico del tumor, asegurar una adecuada aplicación de medicamentos y evitar confusiones en cuanto a qué tipo de medicamento aplicar ante la posibilidad de que el tumor desarrolle resistencia. Estudios clínicos a nivel mundial indican que la terapia molecular dirigida en contra de las mutaciones en BRAF puede ser prometedora. Sin embargo, aún falta mucho por estudiar, ya que cada compuesto anticanceroso dirigido en contra de cualquier componente de la cadena Ras/Raf/MEK/ERK debe probar su utilidad clínica en los pacientes afectados por el cáncer. Por otro lado, la aplicación al mismo tiempo de varios medicamentos para tratar de inhibir la cadena Ras/Raf/MEK/ERK y la combinación con quimioterapia tradicional parece ser el plan más efectivo para el tratamiento del cáncer de tiroides. No obstante, el riesgo de que se desarrolle resistencia siempre estará latente, por lo que el desarrollo de más y mejores productos para el tratamiento y control del cáncer es de alta prioridad, sobre todo, si el desarrollo de estos medicamentos puede permitir su uso más generalizado en la población, de tal forma que sea un hecho común tenerlos de forma rutinaria en los centros hospitalarios para el bienestar de los pacientes afectados de cáncer.
Responsabilidades éticasProtección de personas y animalesLos autores declaran que para esta investigación no se han realizado experimentos en seres humanos ni en animales.
Confidencialidad de los datosLos autores declaran que han seguido los protocolos de su centro de trabajo sobre la publicación de datos de pacientes.
Derecho a la privacidad y consentimiento informadoLos autores declaran que en este artículo no aparecen datos de pacientes.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.
El Dr. Paul Mondragón Terán y el Dr. José Gutiérrez Salinas agradecen el apoyo del «Programa de Investigación Científica y Tecnológica del ISSSTE» (Clave E015). Los autores agradecen al biólogo Miguel Ángel Juárez Mancera por su apoyo en la revisión bibliográfica; al Sr. Sergio Hernández Rodríguez por su apoyo en la edición de figuras y a la Srta. Cinthia Santiago Nicolás (División de Investigación Biomédica, CMN 20 de Noviembre, ISSSTE) por su ayuda en el trabajo secretarial.