




El tumor intraabdominal desmoplásico de células pequeñas y redondas es una entidad rara y agresiva con solo 200 casos reportados, con una incidencia mayor en varones y predilección por la segunda década de la vida. Histológicamente se caracteriza por la presencia de nidos de células tumorales pequeñas e indiferenciadas, envueltas en estroma fibroso y desmoplásico.
Caso clínicoVarón de 24 años, que en las 4 semanas previas comienza con dolor abdominal en hipocondrio derecho, tipo cólico, esporádico, autolimitado, acompañado de saciedad temprana, hiporexia y pérdida involuntaria de 10kg de peso en 3 meses. A su ingreso se encuentra abdomen globoso, con peristalsis disminuida, blando, depresible. La tomografía computada de abdomen evidencia múltiples adenomegalias en la cavidad abdominopélvica. Se realiza laparotomía exploradora con la consecuente resección del omento por la presencia de múltiples tumoraciones, las cuales microscópicamente se caracterizaban por grupos de células redondas, pequeñas y azules separadas por un estroma desmoplásico. La inmunohistoquímica reveló positividad para desmina (>75%), antígeno de membrana epitelial (>75%), CD99 (>50%) y S100 (25%), por lo que se diagnostica tumor desmoplásico abdominal de células redondas, pequeñas y azules. Se inició tratamiento quimioterapéutico con base en esquema IMAP más GM-CSF.
ConclusionesEl tumor intraabdominal desmoplásico de células pequeñas y redondas es un tumor poco frecuente, de complejidad diagnóstica y de curso letal. Clínicamente presenta manifestaciones inespecíficas. Histológicamente se clasifica como un sarcoma agresivo de tejidos blandos, que comparte características similares con la familia de tumores de células pequeñas y azules.
The desmoplastic small round cell tumour is a rare and aggressive intra-abdominal neoplasia, with only 200 cases reported, and a higher incidence in men and predilection for the second decade of life. Histologically characterized by the presence of small nests of undifferentiated tumour cells, wrapped in fibrous desmoplastic stroma.
Clinical caseA 24 year old male started with abdominal pain of 4 weeks onset in the right upper quadrant, colic type, sporadic, self-limiting and accompanied by early satiety, decreased appetite, and involuntary weight loss of 10 kg in 3 months. At the time of admission the abdomen was globular, with decreased peristalsis, soft, depressible. Computed tomography of the abdomen showed multiple enlarged lymph nodes in the abdominal-pelvic cavity. A laparotomy was performed, with a subsequent omentum resection due to the presence of multiple tumours, which microscopically were characterised by groups of small, round, blue cells, separated by a desmoplastic stroma. The immunohistochemistry was positive for desmin (> 75%), epithelial membrane antigen (> 75%), CD99 (> 50%), and S100 (25%), concluding with an abdominal tumour of small, round, blue cells as a diagnosis. Chemotherapy treatment was initiated based on IMAP plus GM-CSF.
ConclusionsThe desmoplastic small round cell tumour is a rare neoplasia, with diagnostic complexity and a lethal course. Its clinical presentation is unspecific. Histologically, it is classified as an aggressive soft tissue sarcoma that shares similar characteristics with the family of the small and blue cells tumours.
El tumor intraabdominal desmoplásico de células pequeñas y redondas es una neoplasia de carácter agresivo, poco frecuente, con aproximadamente 200 casos reportados desde su primera descripción en 1989 por Gerald y Rosai1, que señalaron que presenta una mayor incidencia en el sexo masculino, con una relación de 2-10:1, y una predilección por la segunda década de la vida en el 80% de los casos (rango 4-52 años)1–9.
Afecta predominantemente las membranas serosas, principalmente el peritoneo, de las cuales el 62% de las lesiones se localizan en abdomen y el 36% restante ocurren en pelvis; se encuentran casos de tumor primario localizado en testículo, escroto, pleura, fosa posterior, porción petrosa del hueso temporal, órbita ocular y órganos abdominales. La mayoría de los casos se presentan en estadios avanzados como tumores de gran volumen sin un aparente origen orgánico, que pueden estar acompañados por extensos implantes tumorales a lo largo del peritoneo2–6,8.
Histológicamente se caracteriza por la presencia de nidos de células tumorales pequeñas e indiferenciadas, envueltas de un estroma fibroso y desmoplásico. Se trata de un sarcoma (de células redondas) que genéticamente se caracteriza por la expresión de una traslocación recíproca del gen t(11;22)(p13;q11 o q12), resultado de una fusión del gen del sarcoma de Ewing y el cromosoma 22 del tumor de Wilms (WT1), de localización principalmente intraabdominal2,3,5,6,8–11.
El objetivo del reporte es presentar un caso clásico de un tumor desmoplásico de células pequeñas y redondas.
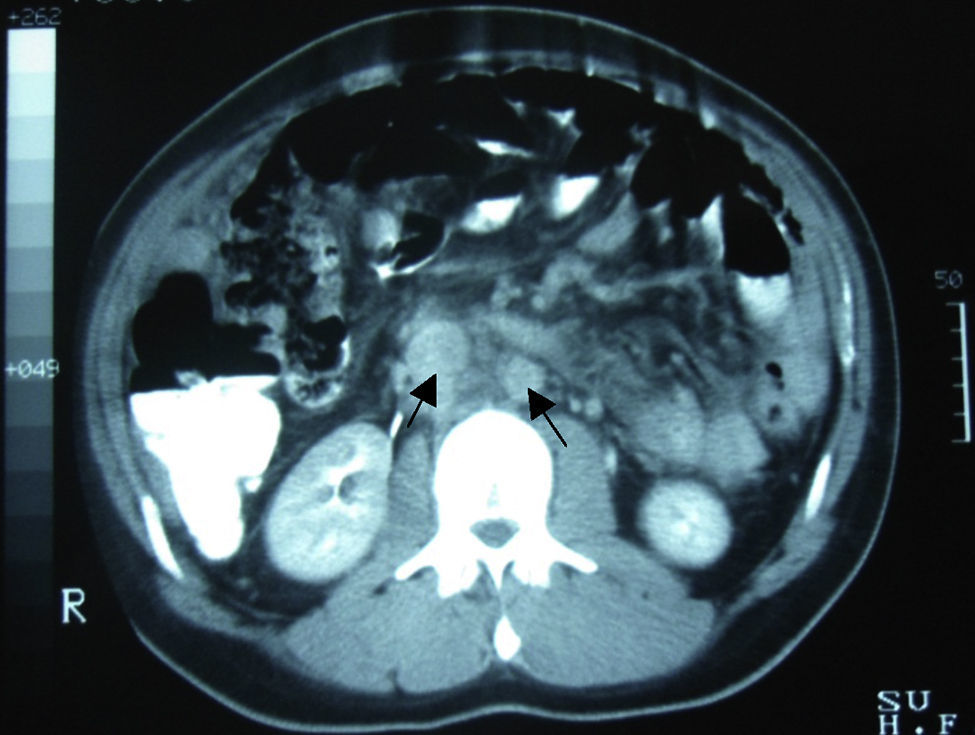
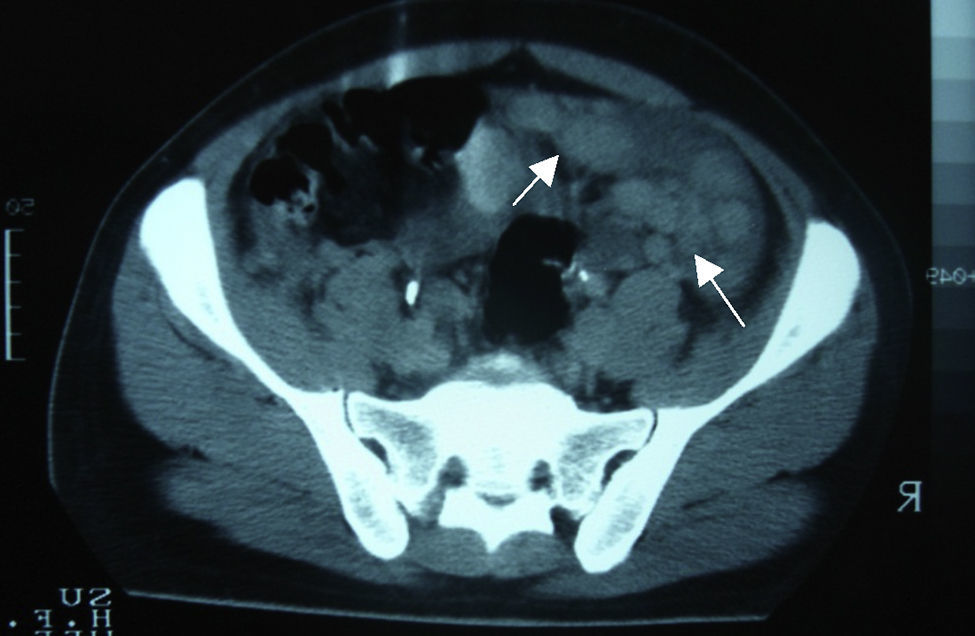
Caso clínicoVarón de 24 años de edad, sin antecedentes quirúrgicos, traumáticos, autoinmunes o ingesta de medicamentos. Inicia cuadro clínico 4 semanas antes de su ingreso con dolor abdominal de inicio súbito en el hipocondrio derecho, de tipo cólico, esporádico, de 10min de duración, acompañado de saciedad temprana, hiporexia y pérdida de 10kg de peso involuntaria en los últimos 3 meses. El paciente niega fiebre, astenia, adinamia, así como otros antecedentes de importancia. A su ingreso se le encontró hemodinámicamente estable, sin fiebre y sin datos patológicos de importancia. Presentaba un abdomen globoso, con peristalsis disminuida, blando, depresible, sin dolor a la palpación y sin visceromegalias. Alfa-fetoproteína de 4.28UI/ml, CA-125 de 110UI/ml y deshidrogenasa láctica de 1,025UI/l. En la tomografía computada de abdomen se evidenció la presencia de múltiples lesiones nodulares a ovoides en la cavidad abdominopélvica, de densidad aumentada, las cuales aparentemente no surgían de ningún órgano, la mayor de 6×4cm, bien delimitadas, hallazgos que evidenciaban carcinomatosis peritoneal con adenopatías retroperitoneales metastásicas (figs. 1 y 2).
 en retroperitoneo, ganglios paraaórticos y abdominales (flechas).")
.")
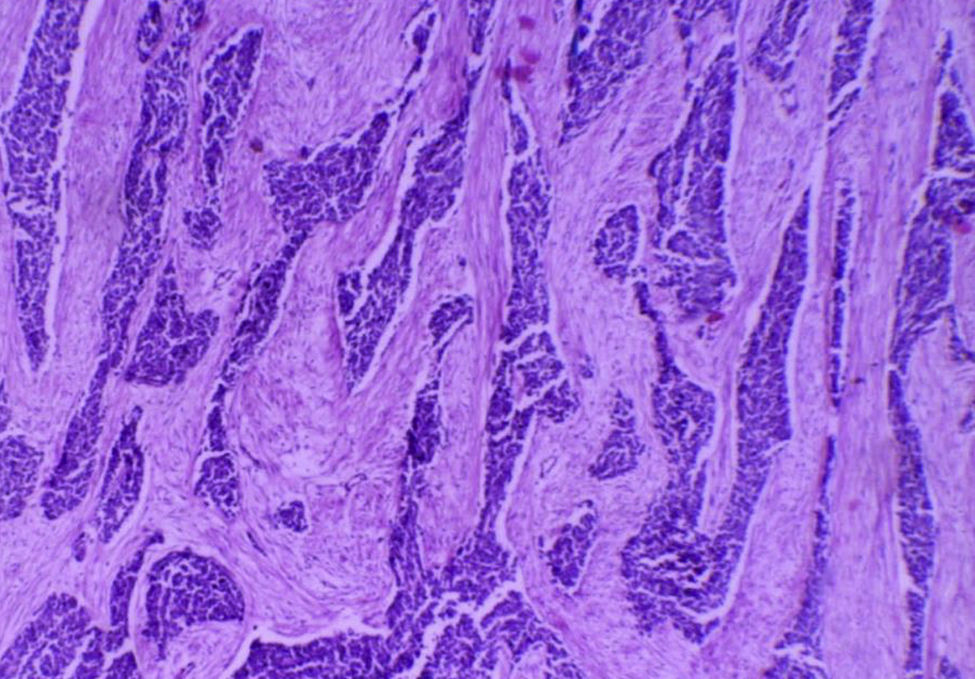
Se realiza laparotomía exploradora, en la cual se puso de manifiesto la presencia de ascitis de aproximadamente 1,000×1cc, que demostró un frotis hemorrágico, hipercelular, constituido por células mesoteliales reactivas, así como la presencia de células de aspecto neoplásico con tendencia a formar estructuras moriformes y papilares con marcada atipia. Se palpó tumoración a nivel de lóbulo hepático derecho de aproximadamente 17×14cm, indurada y de bordes definidos; se evidenció la presencia de adenopatías retroperitoneales, paraaórticas y parailiares, sin embargo, no se biopsió ningún ganglio. No había evidencia de otra extensión macroscópica a diafragmas o estructuras de la pelvis. Se observó la presencia de múltiples nodulaciones sólidas e irregulares localizadas en epiplón, por lo que se procedió a realizar omentectomía total (fig. 3) como tratamiento y diagnóstico quirúrgico, que fueron subóptimos por el estadio avanzado de la enfermedad.

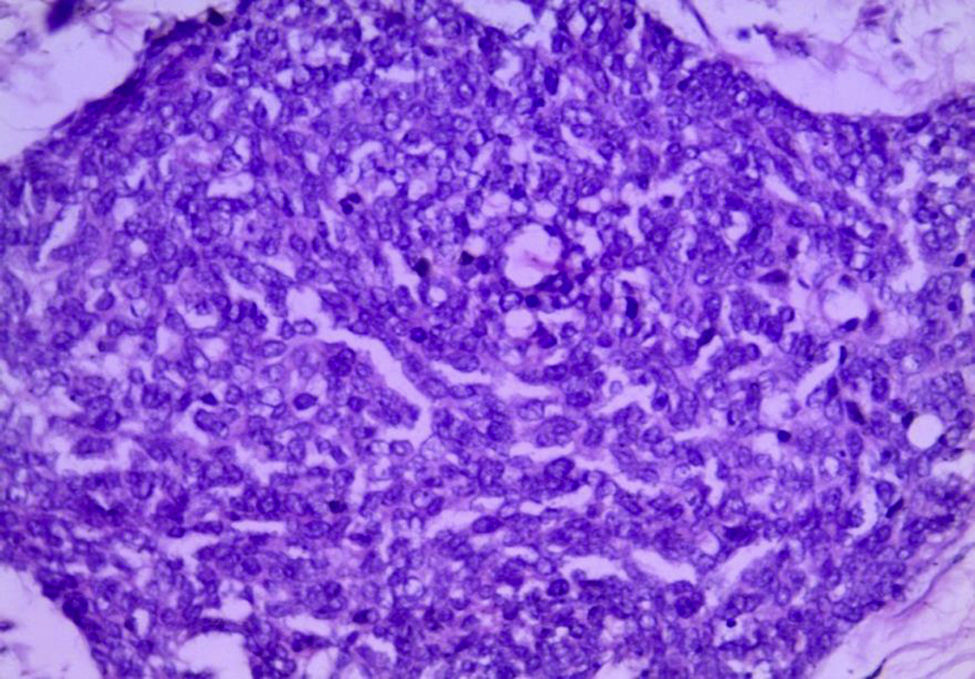
Para el análisis anatomopatológico se obtuvo un espécimen de 52×22×7cm, de color café-violáceo, altamente vascularizado, y que presentaba múltiples tumoraciones nodulares, la mayor de 6×4×4cm y la menor de 0.5×0.5×0.5cm. Los nódulos presentaban una superficie externa lisa y vascularizada, de consistencia firme al corte, superficie homogénea y color blanco-amarillento (fig. 4). Microscópicamente las lesiones mostraron la presencia de grupos de células redondas, pequeñas y azules, las cuales formaban nidos y cordones con áreas sólidas rodeadas por un estroma de aspecto desmoplásico con abundantes vasos sanguíneos y alta permeación linfática; las células eran uniformes, de escaso citoplasma, bordes mal definidos, con abundantes mitosis, dispuestas en forma rosetoide, seudoglandular y en empalizada (figs. 5 y 6). La inmunohistoquímica reveló positividad para desmina (+++o>75%), antígeno de membrana epitelial (+++), citoqueratina (+++), CD99 (++o>50%) y S100 (+o≤25%). Reunía criterios clínicos, histológicos e inmunohistoquímicos para realizar el diagnóstico de tumor abdominal de células redondas, pequeñas y azules.



Se inició tratamiento médico con esquema de quimioterapia IMAP (vincristina, doxurrubicina, ciclofosfamida, isofosfamida y etopósido) más factor estimulante de colonias de granulocitos y macrófagos, que solo se empleó con finalidad paliativa. Durante el decimosegundo ciclo el paciente presentó disnea, epistaxis, pancitopenia y aumento de azoados, por lo que fue necesaria la descontinuación de la quimioterapia por aplasia medular secundaria; 10 días después de dicho curso clínico el paciente falleció.
DiscusiónEl tumor desmoplásico de células redondas y pequeñas es poco frecuente, agresivo, perteneciente a la familia de las neoplasias conocidas como tumores de células pequeñas, redondas y azules, los cuales incluyen linfomas no Hodgkin, neuroblastoma, rabdomiosarcoma, sarcoma de Ewing, tumor de Wilms y tumor neuroectodérmico primitivo periférico2,4,5,9,12,13.
La histogénesis es incierta, pero se ha visto asociada a una relación con el mesotelio, denominándose mesotelioblastoma fundamentado en: frecuente relación con superficies serosas, inmunorreactividad para WT1 y expresión de marcadores epiteliales y mesenquimales2,5,6,11.
La sintomatología más común es la presencia de dolor abdominal, distención y tumores abdominales palpables. El 50% de los casos puede llegar a cursar con clínica compatible de seudoobstrucción intestinal, diaforesis nocturna, pérdida de peso, hematuria y derrame pleural4,12,14.
Aproximadamente el 30% de los casos se presentan con ascitis, que en el estudio citológico muestra una población con pérdida de cohesividad, con células morfológicamente redondas, ovales o espiculadas con escaso citoplasma, membrana nuclear irregular, cromatina granular y nucléolo inconspicuo. Presley et al.12 describieron que la morfología puede modificarse después del tratamiento con quimioterapia, la cual genera pérdida de la cohesividad, cariomegalia y ausencia de mitosis3,6,12.
Dentro de los estudios de gabinete para su diagnóstico la tomografía computada abdominopélvica típicamente se presenta con uno o múltiples tumores de contornos lobulados bien definidos, partes blandas hiperdensas y heterogéneas, con áreas de hipodensidad relacionadas a focos de necrosis y hemorragia, de localización intraperitoneal y sin un aparente origen de un órgano abdominal; el 50% pueden presentar adenomegalias, y el 20%, calcificaciones. Bellah et al.15 reportaron la localización de lesiones en recto vesical y/o recto urinario en un 82%3–5,15.
En la resonancia magnética el tumor se evidencia con múltiples neoplasias intraperitoneales de señal intermedia en secuencias potenciales en T1, realce en la fase T2 y con poca captación del contraste intravenoso5,14. Radiológicamente no se ha demostrado una adecuada especificidad que apoye el diagnóstico, siendo necesaria la correlación clínica/patológica.
Histológicamente presenta un patrón de células pequeñas, redondas, ovales y/o ahusadas, con núcleos hipercromáticos, escaso citoplasma con abundantes mitosis, las cuales se disponen en nidos, cordones, fila india, sábanas, trabéculas y áreas sólidas bien delimitadas, embebidas en un estroma desmoplásico. Existen patrones morfológicos menos frecuentes, como son los papilares, los glandulares y los cribiformes con células claras, fusiformes, pleomórficas, rabdoides y basaloides que forman rosetas o seudorrosetas2,5,6,9,14. En nuestro caso, la disposición celular fue la convencional, así como la formación de rosetas, empalizada periférica y nidos con necrosis central.
Inmunofenotípicamente se caracteriza por presentar un patrón con expresión para marcadores epiteliales, mesenquimales y neuronales. Este tumor presenta positividad para desmina (100%), AE1-AE3 (100%), antígeno de membrana epitelial (100%), citoqueratina (100%) y enolasa neuronal específica (100%); S100 y CD99 se expresan parcialmente positivos2–5,10,11, como fue el caso de nuestro paciente.
En 1991 fue descrito por primera vez genéticamente con una fusión de los genes EWS del sarcoma de Ewing en su exón 7 y el exón 8 del gen WT1 del tumor de Wilms; esta traslocación recíproca caracterizada t(11:22)(p13;q11) o (p13;q12) puede ser detectada mediante PCR-RT con una sensibilidad del 93% y una especificidad del 100%, o por hibridación mediante Southern-Blot con una sensibilidad del 97%2,4–6,8–12.
Las opciones terapéuticas son limitadas, se prefiere realizar citorreducción quirúrgica con una remoción parcial o completa del tumor en forma primaria, la cual solo es posible en el 60% de los casos y se asocia con un mejor pronóstico a largo plazo. Este tipo de tumores ha tenido mejor resultado cuando el tratamiento es combinado. Hassan et al. demostraron una supervivencia del 58% a 3 años en los pacientes que recibieron quimioterapia, radioterapia y cirugía citorreductora, en comparación con aquellos en los que no se llevó a cabo ningún tratamiento (0%). El tratamiento exclusivo con quimioterapia se ha asociado a un alto índice de toxicidad, empeorando el pronóstico y la sobrevida de los pacientes3,5.
Debido a que se trata de un tumor infrecuente, no se encuentra establecido un esquema quimioterapéutico de elección, sin embargo, existen diferentes protocolos recomendados, siendo el IMAP más factor estimulante de colonias de granulocitos y macrófagos el mayormente empleado5,16,17, seguido por otros esquemas como el IMAP11, PAVEP (ciclofosfamida, etopósido, doxorrubicina y cisplatino) y PEVEP (ciclofosfamida, etopósido, epirrubicina y cisplatino)13,17.
La complicación a largo plazo más frecuentemente presentada es la obstrucción intestinal, que ocurre aproximadamente hasta en un tercio de los pacientes. El curso clínico es agresivo, con múltiples recidivas locales y metástasis a distancia en pulmón, hígado y ganglios linfáticos5,6.
La mayoría de los casos se detecta en estadios avanzados, los pacientes presentan una vida media menor a 3 años y menos del 15% de los pacientes presentan una sobrevida a 5 años2–4,6,7.
Dentro del diagnóstico diferencial con base en los estudios de imagen se deben considerar los siguientes: mesotelioma, tumor carcinoide, carcinomatosis peritoneal, leiomiomatosis peritoneal, tumor desmoide intraperitoneal, linfomatosis peritoneal, sarcoma peritoneal y tuberculosis peritoneal4.
Histopatológicamente el diagnóstico diferencial es amplio, debiéndose tomar en cuenta los patrones morfológicos e inmunohistoquímicos; en pacientes pediátricos se incluyen: sarcoma de Ewing, rabdomiosarcoma, neuroblastoma, linfoma de células pequeñas, sarcoma sinovial anaplásico y tumor de Wilms; en cambio, en los adultos es importante considerar: linfomas, carcinoma de células pequeñas, carcinoma de células de Merkel, carcinoma neuroendocrino y mesotelioma, entre otros2,7,13.
ConclusionesEl tumor intraabdominal desmoplásico de células pequeñas y redondas es un tumor poco frecuente, de complejidad diagnóstica y de curso letal. Clínicamente presenta manifestaciones inespecíficas. Histológicamente se clasifica como un sarcoma agresivo de tejidos blandos, que comparte características similares con la familia de tumores de células pequeñas y azules.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.