




Introducción
El feocromocitoma es un tumor infrecuente, cuya incidencia es del 1-2 por 100.000 adultos1, y es responsable del 0,1-1% de los casos de hipertensión arterial curable2. Raramente están asociados con una tumoración adrenal cortical contralateral no funcionante. Tan sólo han sido publicados 4 casos en la bibliografía mundial3,4. Debido a esta rareza casual, se describe un caso de feocromocitoma derecho de gran tamaño asociado a un adenoma cortical no funcionante contralateral.
Caso clínico
Mujer de 52 años, sin antecedentes de interés, que consulta por molestias abdominales difusas de 6 meses de evolución, cefaleas y palpitaciones. La presión arterial (PA) era de 150/80 mmHg y la frecuencia cardíaca de 76. La analítica de rutina, la radiografía torácica y la abdominal, el electrocardiograma y la exploración abdominal no evidenciaron alteraciones.
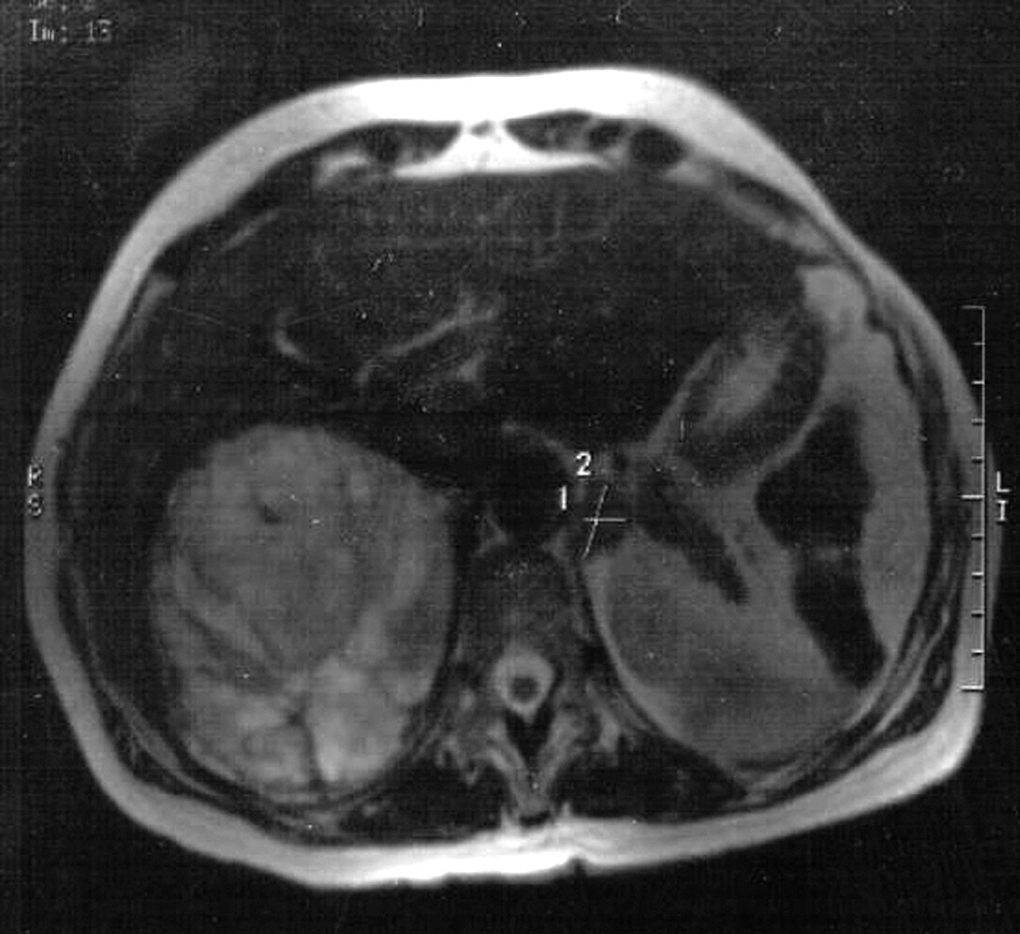
La ecografía abdominal detectó una masa suprarrenal derecha de 12 x 10 cm. La tomografía computarizada (TC) y la resonancia magnética (RM) evidenciaron que se trataba de una masa muy vascularizada, con focos de necrosis y que desplazaba órganos vecinos; también se identificó una lesión en la glándula adrenal izquierda de 1,9 x 1,2 cm sugestiva de adenoma (fig. 1).
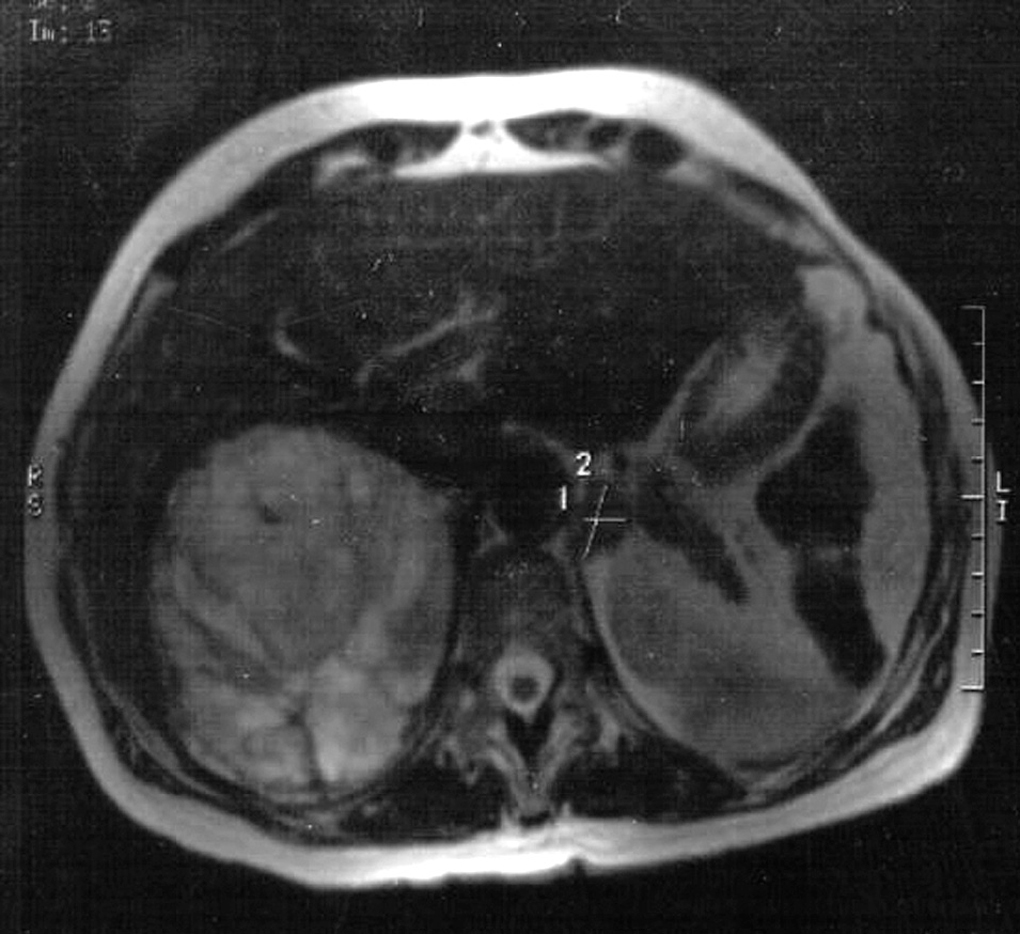
Fig. 1. Resonancia magnética de abdomen, donde se objetiva una gran tumoración adrenal derecha y otra adrenal izquierda de menor tamaño.
El estudio hormonal incluyó la determinación de TSH, T4, ACTH, VIP, FSH, 17-betaestradiol, testosterona, SHBG, andrógeno libre, DHEA-S, cortisol basal y cortisol posdexametasona, y todos ellos presentaron valores normales; sin embargo, el cortisol y las catecolaminas (adrenalina y noradrenalina) en orina se encontraban aumentadas en varias determinaciones.
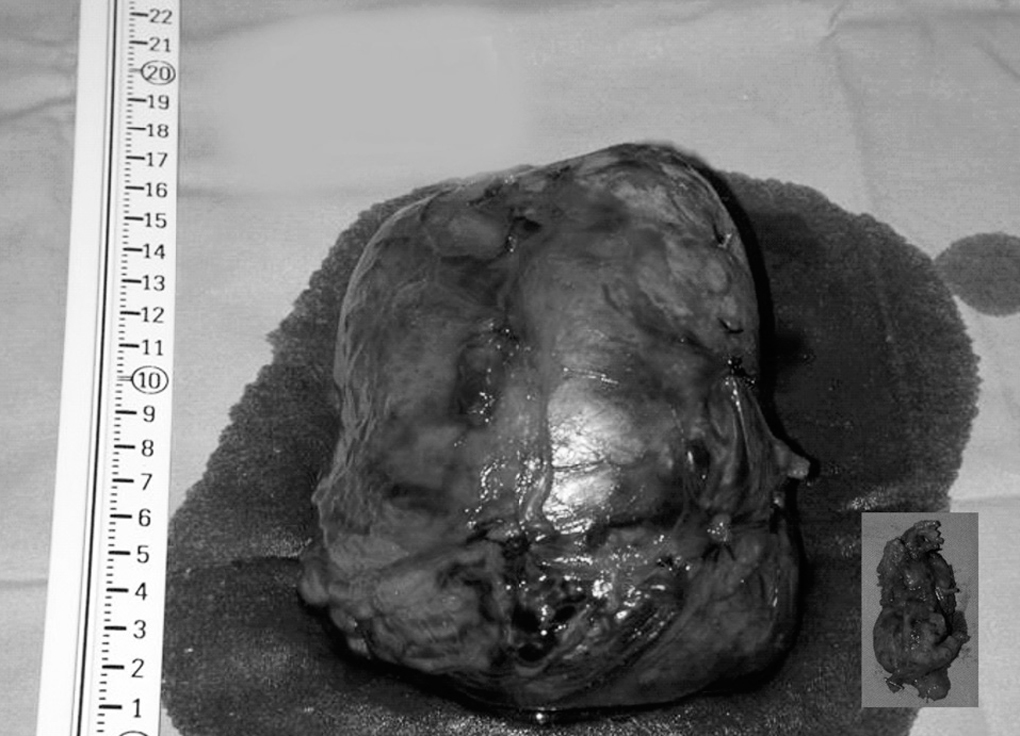
Con la sospecha diagnóstica de tumor maligno suprarrenal derecho, y de adenoma cortical izquierdo frente a nódulo metastásico suprarrenal izquierdo, se decide realizar una intervención quirúrgica, previo bloqueo adrenérgico; se realizó suprarrenalectomía bilateral por vía subcostal bilateral con prolongación torácica derecha (octavo espacio intercostal derecho), y se apreció una tumoración de 14 x 10 cm en la glándula suprarrenal derecha, y un pequeño nódulo de 4 cm en la izquierda (fig. 2).
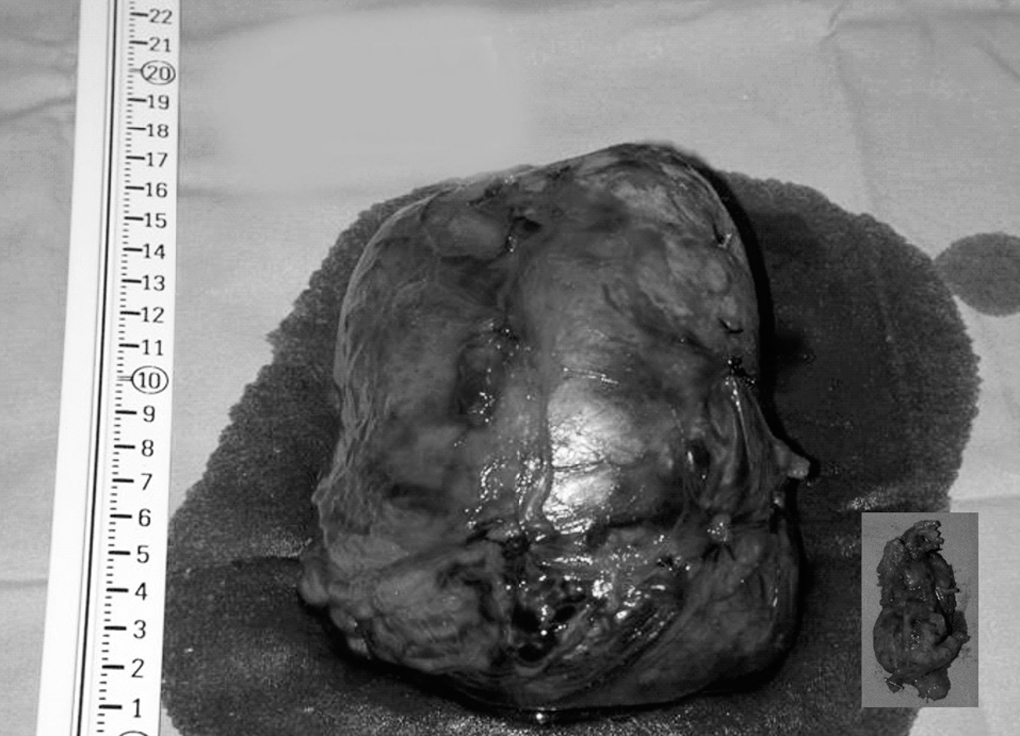
Fig. 2. Fotografía de especímenes quirúrgicos.
El postoperatorio cursó sin complicaciones, y se indicó tratamiento sustitutivo con hidraltesona, con alta al duodécimo día del postoperatorio. Actualmente, la paciente se encuentra asintomática.
El diagnóstico anatomopatológico fue de adenoma cortical izquierdo y de feocromocitoma suprarrenal derecho benigno (tumoración encapsulada, con mitosis muy escasas y con inmunohistoquímica positiva a enolasa, cromogranina y sinaptofisina).
Discusión
Los feocromocitomas son tumores muy poco frecuentes; en los Estados Unidos se diagnostican alrededor de 800 casos al año5. Producen, almacenan y segregan catecolaminas. Se originan generalmente en la médula suprarrenal, aunque un 10% puede desarrollarse a partir de las células cromafines en los ganglios simpáticos del abdomen y tórax, y en el órgano de Zuckerland (feocromocitomas extraadrenales o paragangliomas).
En el adulto, el 80% son unilaterales y el 10% bilaterales, esto es mucho más común en el feocromocitoma familiar (5% de los casos). Menos del 10% sigue un curso evolutivo maligno, el cual viene determinado por la invasión local de tejidos vecinos o la presencia de metástasis a distancia6.
La sintomatología clásica consiste en episodios de cefalea, sudoración y taquicardia, independientemente de las cifras de PA previas. Estas crisis paroxísticas aparecen en el 75% de los casos con cifras elevadas de PA.
El algoritmo de estudio diagnóstico se basa en7: a) diagnóstico bioquímico de certeza: catecolaminas y metabolitos urinarios, y catecolaminas plasmáticas; b) diagnóstico de localización: TC, RM y centellografía con un análogo de la guanetidina (MIBG) marcado con yodo, y c) cateterismo venoso.
La vía de abordaje depende de la localización del tumor. Las más utilizadas son la incisión subcostal derecha o izquierda, o bilateral. Raramente se emplea la vía posterior. El abordaje laparoscópico es otra alternativa que día a día está adquiriendo mayor relevancia. Para los paragangliomas abdominales, la vía de elección es la laparotomía media8.
En general, el tratamiento del feocromocitoma conlleva la escisión quirúrgica del tumor. Es necesario un adecuado manejo preoperatorio que conlleva los bloqueos alfa y betaadrenérgicos, y la utilización de expansores de volumen plasmático9.
La extirpación quirúrgica completa cura la hipertensión en el 75% de los pacientes; en los demás casos, ésta recidiva y habitualmente se controla con las medidas terapéuticas convencionales. La eliminación de catecolaminas se normaliza aproximadamente a la semana de la cirugía. La supervivencia a los 5 años poscirugía suele ser superior al 95% (en los malignos, menor del 50%) y la tasa de recidivas inferior al 10%6.
Los adenomas corticales suprarrenales constituyen una neoplasia benigna de células corticales suprarrenales que originan síntomas por exceso en la secreción hormonal. La resección está indicada ante una producción hormonal excesiva, y el tratamiento definitivo es la extirpación completa de la glándula con el adenoma. El diagnóstico diferencial adenoma/carcinoma es muy difícil basándose en la morfología celular. Los tumores > 6 cm que ocasionan síndromes suprarrenogenitales suelen ser carcinomas.
Los adenomas corticales asintomáticos unilaterales representan la mayoría de los incidentalomas sólidos adrenales10. Los feocromocitomas asintomáticos y de gran tamaño son muy infrecuentes, cuando son bilaterales se suelen asociar a otro feocromocitoma y sólo de forma excepcional se presenta la asociación con un adenoma cortical. Sólo se han publicado en la bibliografía mundial 4 casos de coexistencia de feocromocitoma con adenoma cortical suprarrenal no funcionante3,4.
En la actualidad, la coexistencia de ambas patologías se considera tan sólo una mera coincidencia, al derivar las células cromafines y las células adrenales corticales de diferentes stem-cells; de este modo, un defecto familiar en una línea celular no afecta a la otra3.
El empleo de las modernas técnicas diagnósticas radiológicas permitirá incrementar el número de casos diagnosticados y publicados en el futuro, de cuyo estudio se obtendrá el mecanismo fisiopatológico causal de dicha coexistencia tumoral.
Correspondencia: Inmaculada Monjero Ares.
Servicio de CGDA.
Hospital Xeral. C/ Severo Ochoa, s/n.
27004 Lugo. España.
Correo electrónico: inmamonj@hotmail.com
Manuscrito recibido el 9-12-2004 y aceptado el 31-3-2005.

