




Aunque la mayoría (95%) de las neoplasias mesenquimales gastrointestinales corresponden a tumores del estroma gastrointestinal (GIST) y de músculo liso, también se ha descrito una gran variedad de otros tipos tumorales con muy baja incidencia, tales como schwannoma, tumor desmoide, tumor fibroso solitario, pólipo fibroide inflamatorio, tumor miofibroblástico inflamatorio, etc.1,2. El principal problema diagnóstico que presentan estas lesiones inusuales es su fácil confusión con determinados cuadros morfológicos de GIST, y especialmente con los GIST CD117-negativos y sin mutaciones en los genes KIT o PDGFRA (wtKIT/PDGFRA).
Recientemente, se ha llamado la atención sobre una forma rara de neoplasia gastrointestinal mesenquimal de curso clínico benigno, diferenciación fibro/miofibroblástica y presencia de focos de calcificación, que ha sido denominada con los términos descriptivos de tumor fibroso de la infancia con cuerpos de psammoma3 y seudotumor fibroso calcificante4.
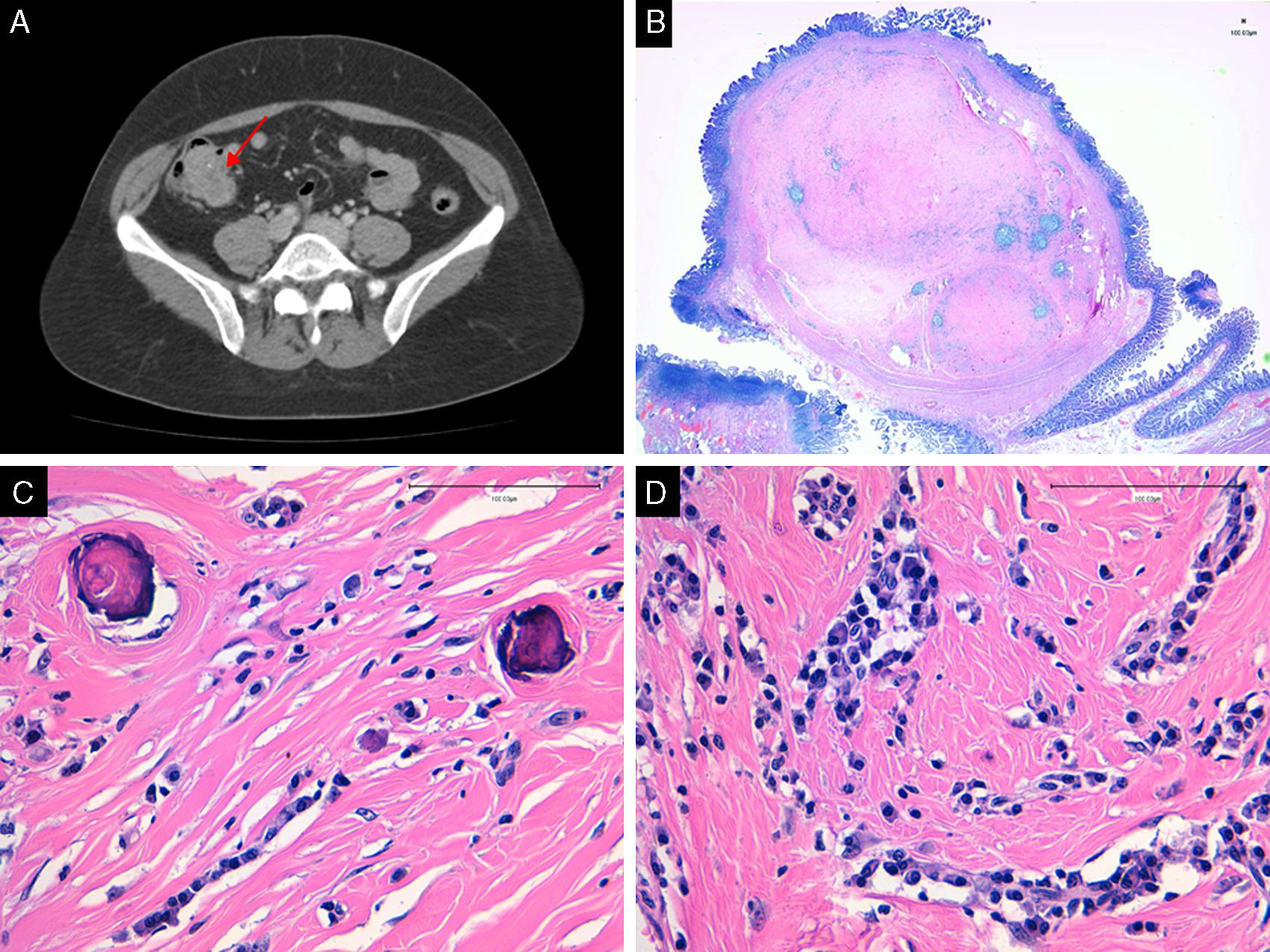
Presentamos el caso de una mujer de 46 años, hipertensa en tratamiento, sin otros antecedentes de interés, que consultó a su médico de atención primaria por dolor abdominal recurrente y náuseas. A la paciente se le realizó una ecografía abdominal por sospecha de litiasis biliar, advirtiéndose engrosamiento de una asa próxima al íleon terminal, con una pared de 5mm de grosor, no dolorosa y líquido en cuantía moderada en pelvis, espacio interasas y saco de Douglas. La colonoscopia puso de manifiesto una lesión submucosa de 2,5×3cm situada en íleon terminal, cuya biopsia fue muy superficial y no diagnóstica. Posteriormente, se realizó una tomografía computarizada con contraste, que reveló una tumoración de densidad de partes blandas, de 4,5×2,8×2,6cm, localizada a nivel de íleon terminal y válvula ileocecal, que protruía en el interior del ciego, de contorno bien definido, con hiperdensidades puntiformes en su interior, compatibles con calcificaciones; no había dilatación de asas intestinales, adenopatías locorregionales, ni afectación hepática o pulmonar (fig. 1 A). Con la sospecha clínica de GIST ileal, se propuso una resección ileocólica, mediante abordaje laparoscópico. La paciente cursó sin complicaciones y fue dada de alta a los 3 días de la intervención.
 Imagen de la TAC de abdomen en la que se observa lesión ileal con calcificaciones. B) Tumor bien delimitado situado en la submucosa. C) La neoplasia muestra calcificaciones tipo psammoma (HE 40X). D) Se observa estroma colágeno denso e infiltrado inflamatorio linfoplasmacitario (HE 40X).")
A) Imagen de la TAC de abdomen en la que se observa lesión ileal con calcificaciones. B) Tumor bien delimitado situado en la submucosa. C) La neoplasia muestra calcificaciones tipo psammoma (HE 40X). D) Se observa estroma colágeno denso e infiltrado inflamatorio linfoplasmacitario (HE 40X).
El estudio anatomopatológico de la pieza quirúrgica puso de manifiesto una lesión bien delimitada y no encapsulada de 2,5cm de dimensión máxima, constituida por una proliferación fibroblástica, hipocelular, que crecía en el seno de una matriz densa, con abundante colágeno. En su seno había escaso infiltrado inflamatorio de células plasmáticas y linfocitos, y focos de calcificación distrófica y cuerpos de psammoma (fig. 1 B-D). Con las técnicas de inmunohistoquímica, las células presentaban inmunorreacción positiva en ocasionales células a CD34 y negativa a CD117, DOG1, S100 y AML. Ante estos hallazgos, se emitió el diagnóstico de tumor fibroso calcificante (TFC) ileal.
El TFC es un tumor de partes blandas, benigno e infrecuente, descrito originalmente en niñas de 2 a 11 años en 1988 por Rosenthal y Abdul-Karim como tumor fibroso de la infancia con cuerpos de psammoma3. Inicialmente, se pensó que representaba un proceso reactivo resultado de una cicatrización anómala, de ahí el nombre de seudotumor fibroso calcificante4. Finalmente, fue denominado TFC en la clasificación de consenso de la Organización Mundial Salud de 2002.
Se han descrito casos de TFC en múltiples localizaciones tales como peritoneo, pleura, mediastino, pulmón, testículo, glándula suprarrenal y otros5-8. Sin embargo, la localización gastrointestinal del TFC es especialmente infrecuente, habiéndose descrito solo alrededor de 10 casos9.
Histopatológicamente, es un tumor circunscrito, no encapsulado, constituido por colágeno hialinizado, células fusiformes sin atipia, infiltrado linfoplasmacitario y cuerpos de psammoma o calcificaciones distróficas10. Suele ser un hallazgo fortuito que se presenta como una formación nodular bien delimitada, tanto en pruebas de imagen como en su posterior estudio histopatológico, y sin capacidad metastatizante. Generalmente, la clínica es muy inespecífica siendo, como en el caso que nos ocupa, dolor abdominal y alteración de hábito intestinal. El diagnóstico diferencial, como tumor fusocelular submucoso, incluye el GIST, el leiomioma o leiomiosarcoma, el schwannoma, el tumor desmoide y el tumor miofibroblástico inflamatorio, entre otros9, y se resuelve fácilmente mediante el estudio inmunohistoquímico.
La rareza del TFC en el tracto gastrointestinal hace que su historia natural sea difícil de delimitar, pero hasta la fecha ninguno ha recidivado o metastatizado. Esto contrasta especialmente con los descritos en otras localizaciones, donde se han descrito recidivas ocasionales4. Por tanto, el tratamiento del TFC gastrointestinal debe ser conservador con seguimiento radiológico, especialmente para las lesiones extirpadas mediante enucleación.
En resumen, pretendemos llamar la atención sobre esta peculiar entidad que fácilmente ha sido confundida con un GIST o un proceso inflamatorio evolucionado.

