




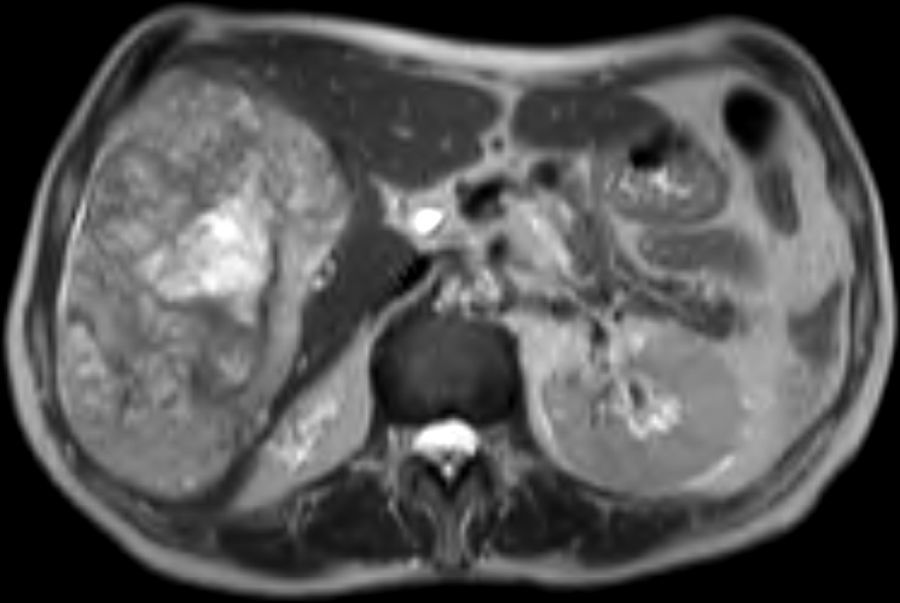
Mujer de 70 años sin antecedentes médico-quirúrgicos de interés que acude a la consulta de Medicina Interna por presentar dolor y sensación de masa en hipocondrio derecho. En la analítica realizada en ese momento no existen alteraciones en el hemograma ni en la bioquímica. Se realiza una TC abdomino-pélvica en el que se describe la existencia de una masa hepática que ocupa la totalidad del lóbulo hepático derecho irrigada fundamentalmente por ramas portales, compatible con un hemangioma gigante. La paciente es remitida a nuestra Unidad de Cirugía Hepatobiliar. Solicitamos una resonancia hepática para una mejor caracterización de la tumoración, para valorar la relación con las diferentes estructuras vasculares y para planificar la técnica quirúrgica. La paciente a las 48 h de la realización de la resonancia hepática acude al Servicio de Urgencias por presentar un cuadro de dolor localizado en epigastrio y síncope. A su llegada se encuentra taquicárdica (FC 130), hipotensa (TA 70-40) y taquipneica (FR 40), en la analítica de sangre se evidencia una anemia franca (Hb 5,8), una trombocitopenia severa (plaquetas 42.000), y una alteración de la coagulación (INR 2,1). La paciente se estabiliza hemodinámicamente mediante el aporte de volumen y mediante la transfusión de 6 concentrados de hematíes, se corrigen las alteraciones de la coagulación mediante la transfusión de 4 pools de plaquetas, y 2.000ml de plasma fresco. En este momento valoramos la resonancia magnética (fig. 1) realizada previamente en la que se describe la existencia de una voluminosa lesión hepática de 18×12cm en lóbulo hepático derecho con sangrado intralesional.

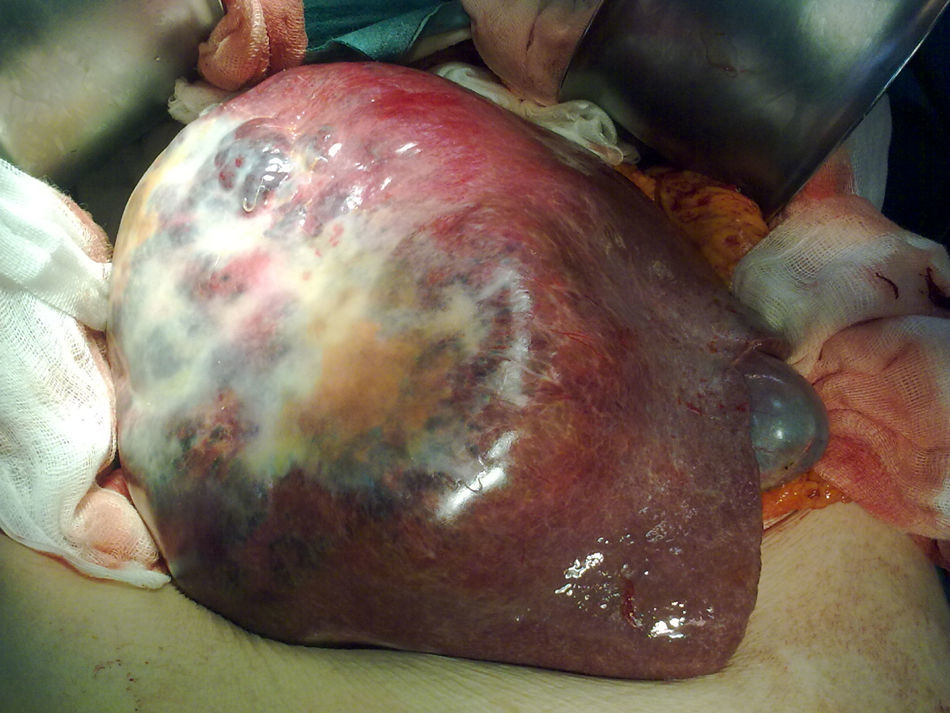
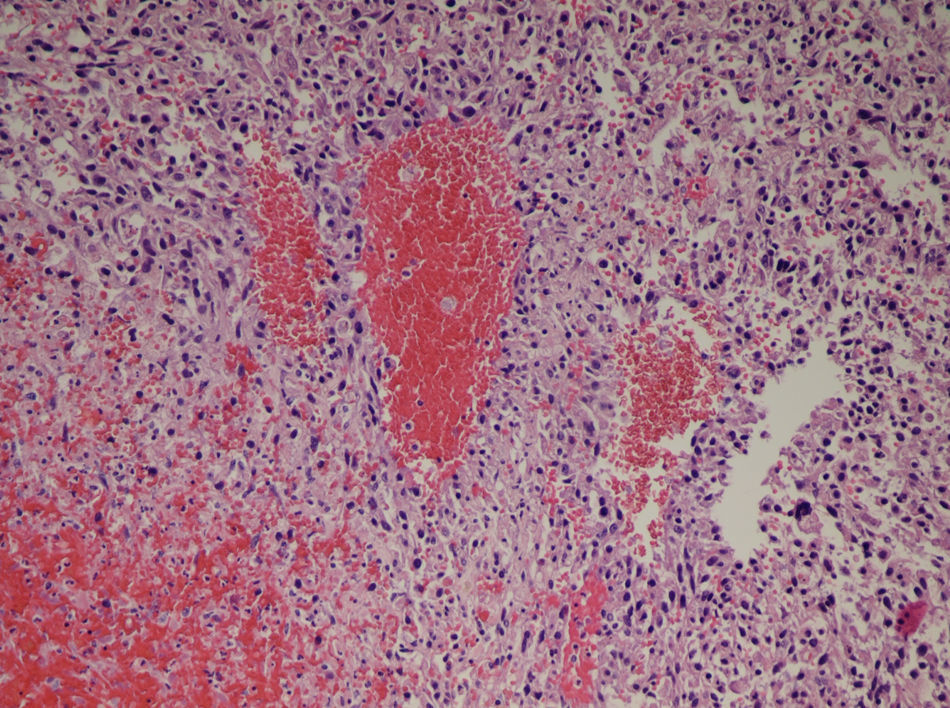
La paciente es valorada por el Servicio de Radiología Intervencionista de nuestro hospital: realizan una ecografía abdominal en la cual no existe líquido libre intraabdominal y descartan la posibilidad de realizar una arteriografía y embolización de la arteria hepática derecha, debido a que el sangrado intratumoral es subsidiario de la rama portal derecha. Trasladamos a la paciente a quirófano, realizamos una laparotomía subcostal derecha ampliada a la línea media y observamos una gran tumoración que ocupa todo el lóbulo hepático derecho desde la cúpula diafragmática derecha hasta la pelvis, de consistencia elástica y de coloración violácea (fig. 2). Realizamos una hepatectomía derecha bajo exclusión vascular total. La paciente es dada de alta al octavo día postoperatorio sin complicaciones. En el estudio anatomo-patológico de la pieza se observa una proliferación celular neoplásica de células epitelioides o fusiformes con marcada atipia celular que forman nidos o estructuras vasculares entremezcladas con áreas de hemorragia y necrosis, mostrando captación celular con marcadores vasculares (CD31, CD34, F.VIII), compatible con angiosarcoma de alto grado (fig. 3).

, compatible con angiosarcoma de alto grado.")
El estudio microscópico de la pieza revela una proliferación celular neoplásica de células epitelioides con marcada atipia celular que forman nidos o estructuras vasculares entremezcladas con áreas de hemorragia y necrosis, mostrando captación celular con marcadores vasculares (CD31, CD34, F.VIII), compatible con angiosarcoma de alto grado.
El angiosarcoma hepático o sarcoma de células de Kupfer es un tumor maligno de origen mesenquimal y de muy baja incidencia (0,14-0,25 por millón de habitantes) y corresponde a un 1,8% de todos lo tumores hepáticos primarios; la edad de presentación se sitúa entre la sexta y séptima décadas de la vida1. Se asocia a la exposición al thorotrast (contraste radiológico empleado hasta la década de los sesenta), al cloruro de vinilo (la serie más amplia de angiosarcoma hepático se publicó en Gran Bretaña en los años 80 y se incluyeron 55 pacientes durante un periodo de 20 años, todos ellos con exposición previa al cloruro de vinilo, empleado para la fabricación de plástico)2, al arsénico (tratamiento en la psoriasis), y a enfermedades como la hemocromatosis y a la enfermedad de Von Reklinghausen. Debido a sus características hipervasculares es difícil diferenciarlo de otros tumores vasculares hepáticos como el hemangioendotelioma, el hepatocarcinoma o el adenoma3. La supervivencia sin tratamiento es inferior a los 6 meses debido a su rápida progresión, a su alto índice de recurrencia y a la falta derespuesta al tratamiento con radio- y quimioterapia. Incluso el trasplante hepático no tiene ningún beneficio debido a su alto índice de recurrencia y a su baja supervivencia tras el trasplante. La resección quirúrgica con margen continúa siendo la única opción de tratamiento, pero es técnicamente muy compleja y en muchas ocasiones la enfermedad está diseminada en el momento del diagnóstico, siendo la resección imposible. Debido a su baja incidencia no está claro, ni existe, ningún protocolo de actuación para el tratamiento del angiosarcoma hepático con sangrado activo4.
En 1940, Kasabach y Merrit describieron un nuevo síndrome en un niño con un hemangioendotelioma kaposiforme, trombocitopenia severa, anemia y coagulopatía de consumo en relación a una activación de la coagulación intravascular en las lesiones vasculares intratumorales lo que conlleva una activación de los factores de la coagulación y de la agregación plaquetaria5. A pesar de que dicho síndrome nunca se haya relacionado con angiosarcomas hepáticos en adultos, existe un claro paralelismo con el caso clínico que hemos descrito previamente en cuanto a la sintomatología clínica, forma de presentación y fisiopatología.

