




Malignant paragangliomas are rare, but may occur especially in patients with familial forms of the disease. We present the case of a 23-year-old woman diagnosed with bilateral carotid paraganglioma with distant and local metastases, associated to a family history of paraganglioma and we present a literature review.
Los paragangliomas de comportamiento maligno son poco frecuentes, pero pueden ocurrir especialmente en pacientes con presentación familiar de la enfermedad. Presentamos el caso de una mujer de 23 años con diagnóstico de glomus carotídeo bilateral con metástasis ganglionares locales y a distancia, asociado a historia familiar de paraganglioma y presentamos una revisión de la literatura.
Carotid body tumours (CBT) are rare. Their reported frequency varies from 1 per 30000 to 100000 individuals in the general population.1 They represent 0.6% of head and neck tumours2 and 50%–60% of paragangliomas (PG) in the whole organism.3,4 Their development is associated with states of chronic hypoxia.5 The main known risk factor in patients with these tumours is that they live in areas higher than 2000m above sea level.6 In 10%–30% of cases there is a family history6,7 of the disease, with dominant autosomal transmission linked to the male sex.3,5 A malignant presentation is reported in 6% of cases, and is strongly associated with cases in the same family.8 The sole definitive criterion for malignancy is the presence of metastasis in local lymphatic ganglia or remote metastasis.9 We present the case of a 23-year-old woman diagnosed with bilateral CBT with ganglion metastasis associated with a family history of PG.
Clinical CaseWe present a 23-year-old woman with a tumour in the left side of the neck and a family history in which her maternal grandmother had CBT. Her condition began 10 years earlier, when she was treated in another institution for a tumour with similar characteristics and location to those of her grandmother. It measured approximately 2cm along its longest axis, was not painful and with an indolent growth pattern, with no associated symptoms. Computerised axial tomography was performed on her neck with a diagnosis of left perithyroidal PG. The lesion was removed surgically by ipsilateral hemithyroidectomy, with no complications. Histopathological diagnosis was of PG measuring 2.3cm along its longest axis, with infiltration into soft tissues and the left lobe of the thyroid gland, immunohistochemically positive for vimentin, chromogranin, synaptophysin, and S-100 protein. Cytokeratin AE1/3 and EMA were negative. Two years later, follow-up using computerised axial tomography detected a left jugular tympanic PG. This was treated by radiotherapy (RT) and embolisation. It evolved satisfactorily until 4 years later, when at the age of 19 she presented in her follow-up with pain in the left side of the neck. Physical examination palpated a nodule in the left neck that magnetic resonance showed to be left carotid PG with tumour activity close to the pyriform fossa. In a third surgical operation the tumour was resected and a covered stent was inserted into the common left carotid artery. Pathology reported a tumour measuring 4cm×1.5cm×1cm composed of nests of cells with broad eosinophilic cytoplasm, with granular chromatin nuclei and occasional nucleoli. In immunohistochemical study the principle cells were chromogranin-positive, while supporting cells were positive for S-100 protein. The histopathological diagnosis was PG.
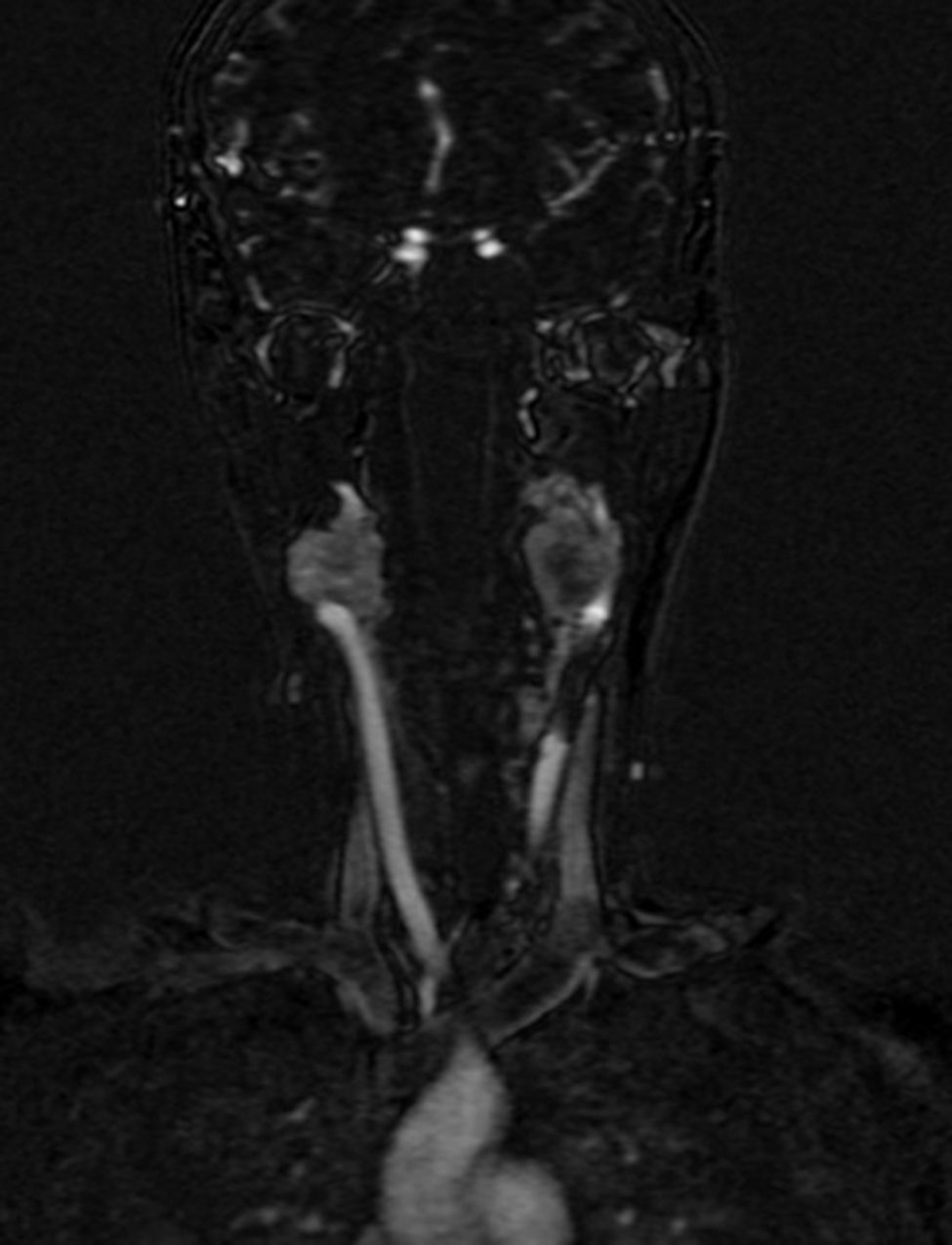
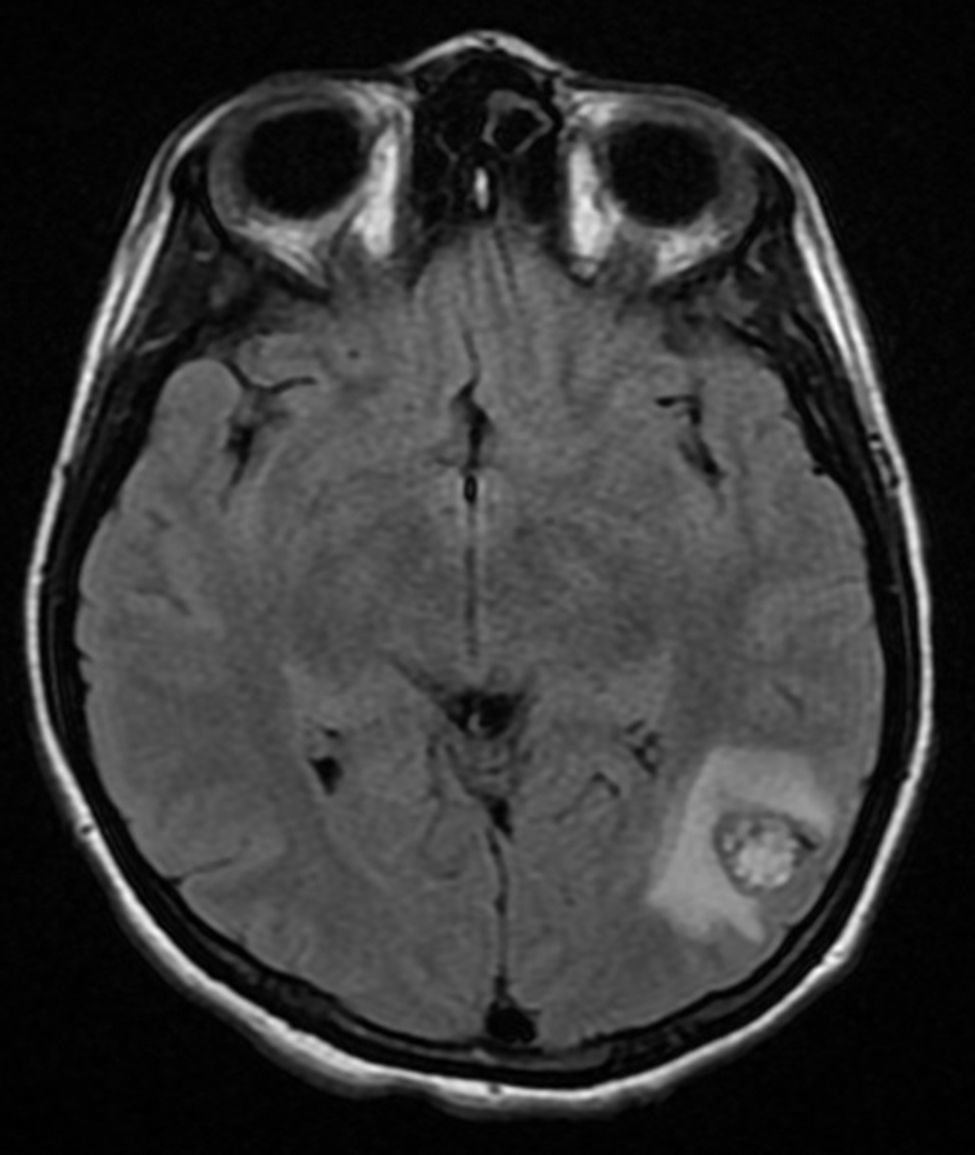
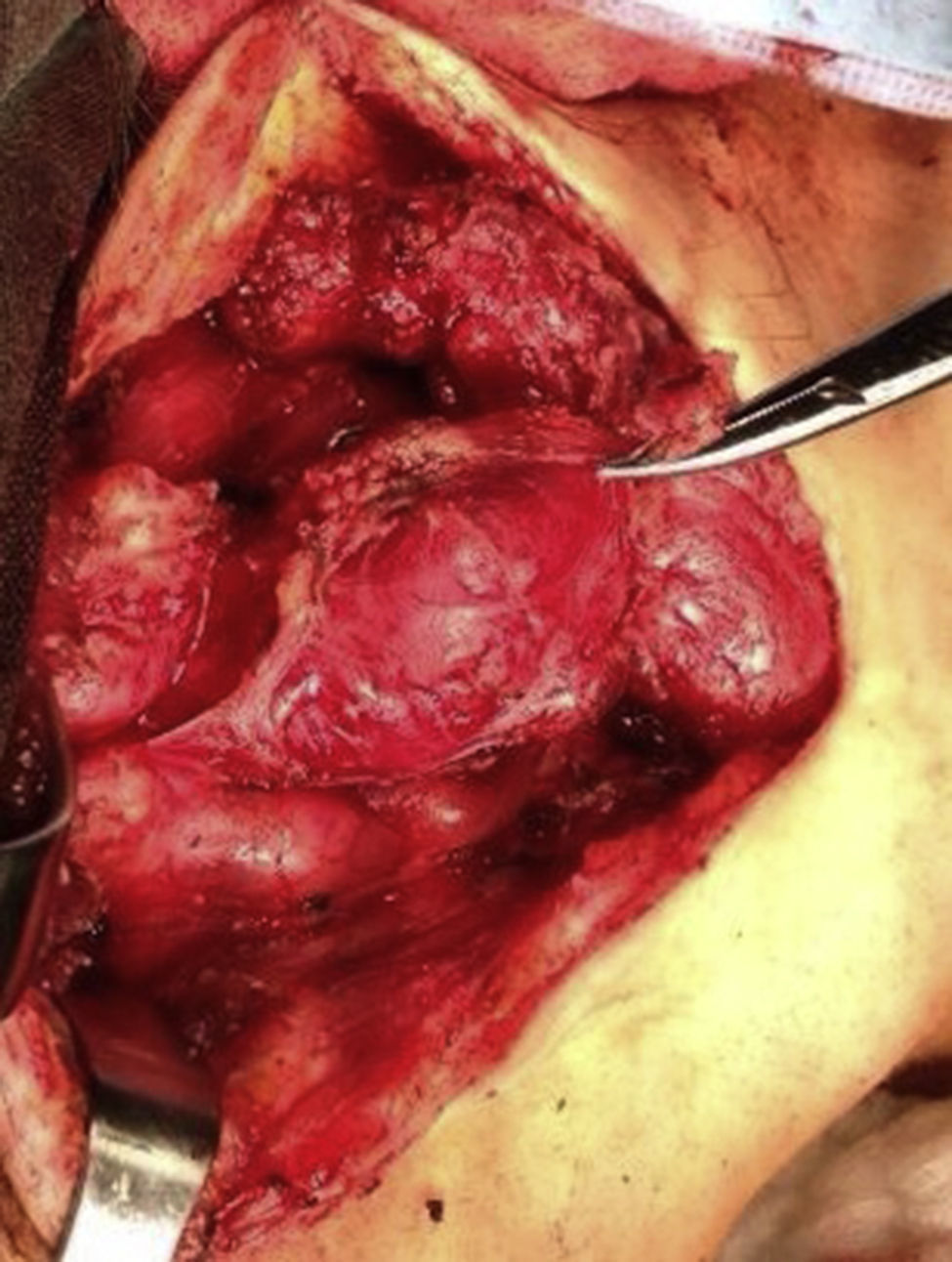
Four years later, at the age of 23, she presented in our department with a recurring tumour in the left side of the neck and left facial hemiparesis. Magnetic resonance showed a 3.5cm left carotid PG extending to the base of the cranium and involvement of the ipsilateral facial nerve, associated with a new right carotid nodule measuring 1.5cm (Fig. 1). Additionally, cerebral images showed a nodular lesion measuring 14mm×14mm×13mm in the second and third left temporal gyri associated with an infratemporal lesion extending to the jugular foramen and the ipsilateral middle ear (Fig. 2). Once again the tumour was resected in the left cervical regions II and III. The tumour was visualised over the carotid sheath (Fig. 3), which was dissected circumferentially without finding any dependent vascular structures. A tumour was found next to the mastoid, and this too was resected. Hypertrophic ganglia were found in the carotid sheath, and these were resected and sent for histopathological study. Pathology received a total of 22 lymphatic ganglia, 3 of which contained metastatic neoplasic cells arranged in a «zellballen» type pattern, with cells of broad eosinophilic cytoplasm and ovoid nuclei. In immunohistochemical study the neoplastic cells expressed chromogranin and S-100 protein with a proliferation index measured using Ki67 of from 5% to 8% (Fig. 4). The largest lymphatic ganglia with metastasis measured 3.5cm×3cm×1cm. The patient evolved with improvement of the facial paralysis and continued treatment with rehabilitation and oral prednisone. The decision was made not to send the patient to RT again, given that the surgical excision was considered to be complete, and as she had already received RT in the same location it was preferred not to exceed the maximum recommended dose. At the time of sending this paper, the patient has now completed 2 years of follow-up free of any recurrent local disease in the left side of the neck.



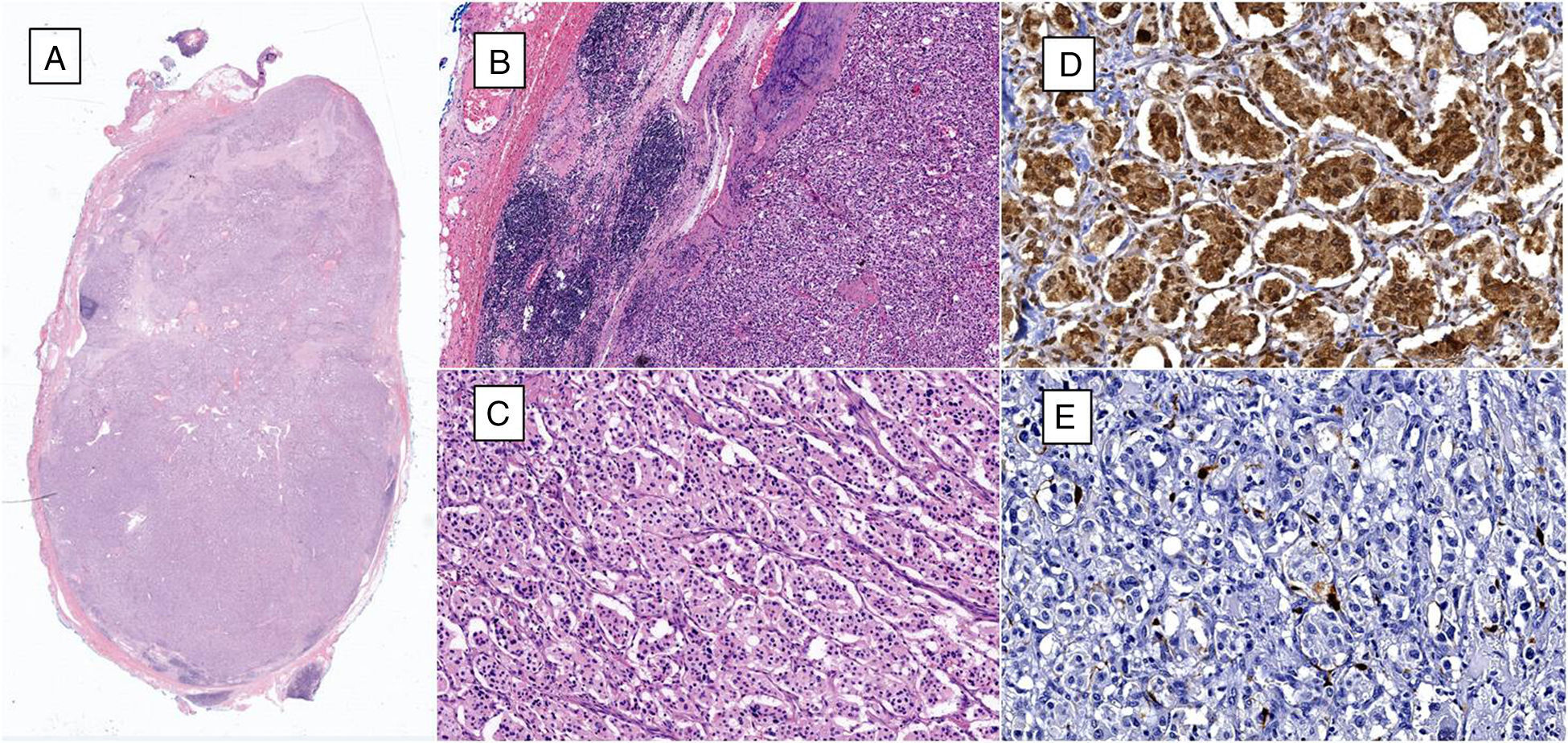
 Microphotograph of lymphatic ganglia containing metastatic neoplastic cells that replace more than 90% of the surface (haematoxylin and eosin, 10×). (B) Residual lymphatic ganglia is identified on the left, while on the right metastatic neoplastic cells can be seen (haematoxylin and eosin, 40×). (C) The neoplastic cells are arranged in a «zellballen» pattern and are composed of broad eosinophilic cytoplasm, with ovoid nuclei and granular chromatin (haematoxylin and eosin, 200×). (D) Immunohistochemistry shows the principal cells to be chromogranin A positive. (E) Immunohistochemistry shows the supporting cells to stand out with S-100 protein.")
(A) Microphotograph of lymphatic ganglia containing metastatic neoplastic cells that replace more than 90% of the surface (haematoxylin and eosin, 10×). (B) Residual lymphatic ganglia is identified on the left, while on the right metastatic neoplastic cells can be seen (haematoxylin and eosin, 40×). (C) The neoplastic cells are arranged in a «zellballen» pattern and are composed of broad eosinophilic cytoplasm, with ovoid nuclei and granular chromatin (haematoxylin and eosin, 200×). (D) Immunohistochemistry shows the principal cells to be chromogranin A positive. (E) Immunohistochemistry shows the supporting cells to stand out with S-100 protein.
PG in the head or neck is rare, but hereditary or metastatic cases are even more so. Correctly categorising these patients makes it possible to select the best form of treatment. Purposefully investigating family history, especially when cases are bilateral or present metastatic disease, will make it possible to choose treatments that tend to prevent recurrence and the possibility of disabling surgical complications, especially those involving cranial nerves.
CBT presents sporadically in 70%–90% of cases and runs in families in 10%–30%, with dominant autosomal transmission linked to the male sex.1,3,5–7 Malign development is reported in 6% of cases of PG of the head and neck, as it is in 2.5%–5% of CBT, chiefly those cases associated with a family history and a tendency towards multiple locations, which are detected due to localised symptoms of the tumour.2,4,8,10 The definitive criterion of malignancy is metastasis to regional lymphatic ganglia or distant organs.9,11 Unlike sporadic cases, family cases present in young patients and are usually in multiple locations as well as bilateral.1,3,7,8 Genetic study is recommended in these patients, given that they are associated with the presence of mutations in the von Hippel–Lindau (VHL) genes, the RET (MEN II) gene, the succinate dehydrogenase (SDH) gene and type I neurofibromatosis in 10%–50% of cases.3,4 In separate studies, Astuti et al. and Bayle et al. determined the role of the SDHD, SDHC, and SDHB deletion, located in chromosome 1q23.3 as the cause of family development of PG.12,13 King et al. showed that SDHB mutation is the main risk factor for the development of metastatic disease at an early age, so it is recommended that early tests be made for SDHD gene mutation in PG of the head or neck.13 In a retrospective study of 59 patients, Lee et al. reported that cases of malign CBT have a higher rate of regional metastasis, with a survival rate at 5 years of 76.3%. Those patients treated using resection of the tumour together with postoperative RT have a better prognosis. His series of reported cases of malignant PG from 1985 to 1996 included only 9 such cases.8
Histologically PG is characterised by a pattern of growth in nests known as «zellballen» (from the German for «cell balls »),6,8,9 which are mainly composed of 2 types of cell: principal cells and supporting cells.1,3 The principal cells produce medium to large catecholamines of the granular eosinophilic cytoplasm and nuclei which vary in size. Immunohistochemistry shows them to be positive for chromogranin A, synaptophysin, and neuron-specific enolase. The supporting cells are elongated cells that surround the principal cells, and histochemistry shows that they express S-100 protein.14,15
Surgery is the treatment of choice in symptomatic cases with accelerated growth and presentation in young patients with small tumours.3,14 It is suggested that surgery be avoided in bilateral or jugular presentations, in patients over the age of 60 and those with arteriosclerosis (except for those with a suspicion of malignancy).3 The German series of Schneider et al.16 concluded that surgical excision is the treatment of choice for small, unilateral, malignant or functional PG. Nevertheless, this same series of 6 patients reported permanent unilateral paralysis of the laryngeal, glossopharyngeal, and hypopharyngeal nerves following resection of a recurring unilateral tumour. Given adverse surgical events involving lesion of the cranial nerves, especially in complicated or recurrent cases, RT has become more popular. With RT there is a high probability of being able to control these tumours at a minimum of risk for the patients, making it an attractive alternative in more complicated cases. The aim of this treatment is to resolve the associated symptoms while preventing progression of the lesion. Stereotactic radiosurgery (Gamma-Knife) is a RT technique which includes a stereotactically guided dose of radiation aimed at a single target.17 A recently reported German series18 followed up 44 patients over 10 years. In 34 patients the size of the tumour was seen to decrease. The dose of RT used was from 10 to 30Gy, with an average of 20Gy. A second series of RT with Gamma-Knife reported follow-up treatment at 14PG. Only 2 of the patients treated had received previous surgery. Doses of from 21.6 to 26.3Gy were used, and patients were followed-up for an average of 40.3 months. Fourteen tumours presented significant regression after treatment, and in 13 of these 14 cases it was possible to preserve the cranial nerves. It was therefore concluded that Gamma-Knife radiosurgery is a good alternative for initial or adjuvant treatment in patients who are not candidates for complete resection.19 In a series of patients with head and neck PG local control of the tumour was shown to occur in 73% of cases, with follow-up at 25 years for those who solely received RT, in comparison with local control in 54% of cases 15 years after treatment for those who only received surgical treatment.15 Radiation affects blood vessels and connective tissue above all, more than it does the tumour cells themselves. Nevertheless, the vascular mass may persist, even after treatment, without this indicating lack of response of the disease.14
Conventional external RT at a dose of 45Gy in 25 fractions is usually sufficient to control the majority of cases of PG.20 It is recommended that the dose of RT should not be exceeded, given that one of the main complications that arise is xerostomia. The flow of saliva is markedly reduced at doses over 10–15Gy, and doses above 50Gy cause irreversible damage to both parotid glands, leading to permanent xerostomia.21 Only one case of paediatric RT has been reported to date, which may be due to the fact that these patients have small tumours that are excellent candidates for surgical treatment.2
Observation is another acceptable decision for some patients, if their underlying medical conditions make it impossible for them to undergo surgical procedures or tolerate the side effects of radiation. This is because the growth of PG is slow and its concomitant negative effects can be tolerated very well for long periods of time. There are some isolated cases of prolonged survival without treatment. Controversy arises because those who favour surgical treatment emphasise the risks of RT, which may induce cerebral necrosis and favours the genesis of other types of tumour, while those who prefer RT as the primary treatment cite the high risk of secondary infection in the cranial wall, as well as cerebrospinal fluid fistulas and mortality associated with surgery.17
Preoperative embolisation has been controversial since it was first described in 1980.1,2,7,22–24 Some authors support the selective embolisation of the vessels which feed the tumour prior to surgical resection. This has the aim of reducing intraoperative bleeding and increasing differentiation of the structures which are involved.1,2,22–24 In spite of this, reporting of cranial nerve involvement varies from one publication to another.7,23 The transarterial approach has the disadvantage that these tumours are usually irrigated by branches which are too small to be catheterised, so that the current tendency is to directly embolise them within the lesion using liquid agents.24 However, this technique is not free of complications, such as central ischaemic compromise.1,22
In cases involving several locations and metastasis, such as ours, it may prove hard to select the best treatment due to the risk involved in resecting the primary tumour and the unknown prognosis of the metastasis.25 In a review of 113 cases, Moskovic et al. concluded that the optimum treatment is broad resection of the tumour with lymphadectomy and postoperative RT, with the best prognosis for patients under the age of 40 years.26 It is recommended that regional lymph nodes be resected, especially when there is a suspicion of metastasis in imaging studies.8
The resection of CBT in general is associated with a 35% morbidity rate and less than 2% intraoperative mortality.5,27 PG metastatic patient survival varies from 1 to 25 years. It is important to follow-up these patients, given their high risk of developing metachronic or synchronic tumours after initial treatment, especially those with family forms of the disease.2,3,7,28
To conclude, it is only possible to define PG as malignant in cases of metastasis at a distance or regional lymphatic ganglia involvement, especially in cases with a family history of PG. Surgery with or without prior embolisation and initial RT with or without surgery are acceptable as treatments of choice. Long-term follow-up is recommended due to the high risk of disease recurrence.
Conflict of InterestsThe authors have no conflict of interests to declare.
Please cite this article as: Cobos González E, Aragón López JA, Soria Céspedes DR, de la Rosa Abaroa MA, Martínez de la Vega Celorio A, Granados Gracia M, et al. Paraganglioma maligno (múltiple, multicéntrico y metastásico) en una paciente con historia familiar de paraganglioma. Cir Esp. 2015;93:e127–e132.
