




Malignant rhabdoid tumors are highly aggressive neoplasms that were initially described in the kidneys of children as a rare variation of Wilms tumors with a rhabdomyosarcomatoid pattern and particularly poor prognosis.1 Subsequently, tumors with histologically similar characteristics were found in other locations, and classified as extrarenal malignant rhabdoid tumors (EMRT).2,3 These neoplasms are rare and have a very aggressive behavior. Their origin has been debated, as they have been described in several solid organs. To date, only 43 cases have been reported in the gastrointestinal tract, 5 the esophagus, 16 the stomach, 10 the small bowel and 12 the colon.4
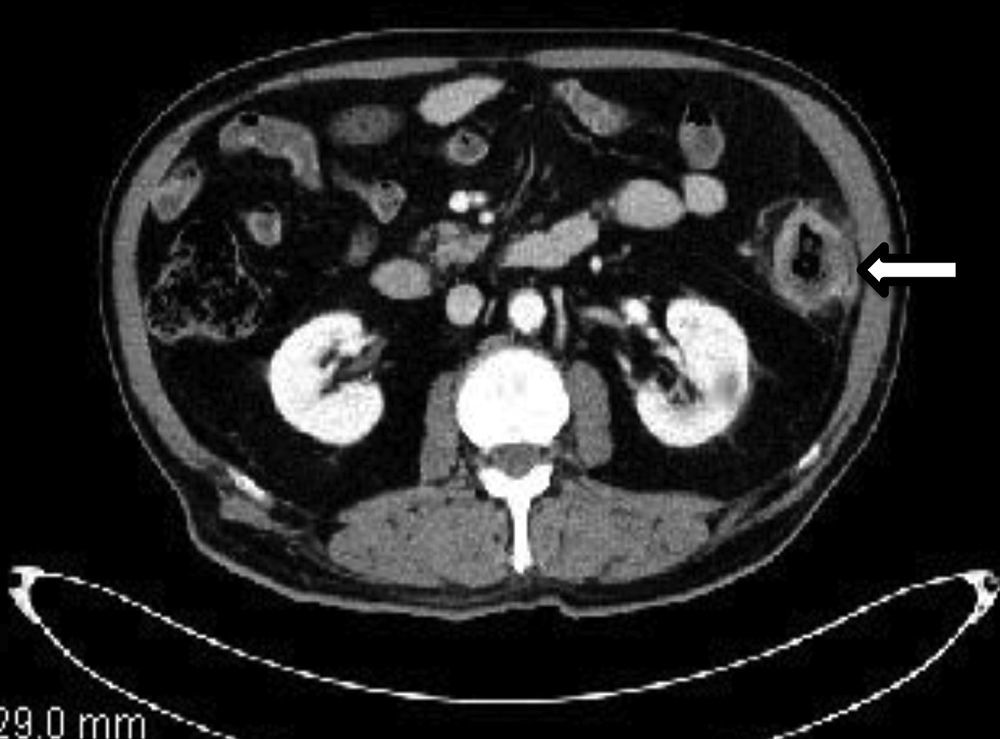
We present the case of a 77-year-old male with a prior history of ischemic cardiopathy who reported having abdominal pain and rectal bleeding over the course of the previous month. Colonoscopy revealed a stenosing neoformation with partial necrosis that was 78cm from the anal margin. Biopsy demonstrated the presence of atypical cells compatible with carcinoma, although immunohistochemistry showed negativity of the tumor cells for CDx-2 and CK 20, which suggested a non-colonic origin. Pre-operative CT showed no evidence of distant disease (Fig. 1). During surgery, a large tumor was found in the descending colon that infiltrated the omentum and parietal peritoneum; a left hemicolectomy with primary anastomosis and resection of the affected abdominal wall was performed. The pathology study determined the lesion was a high-grade infiltrating malignant neoplasm (pT4aN1bMx), with co-expression of cytokeratin cocktail and vimentin (Fig. 2A and B), with negative CK7 and CK20, CDx2 (−), ALC (−), desmin (−), BerEP4 (−), p53 (−), CD117 (−) and calretinin (−), compatible with primary rhabdoid tumor of the colon. On the 7th day post-op, the patient needed a reoperation due to anastomotic leak, which included resection of the anastomosis and terminal colostomy in the left lower quadrant. The patient's postoperative recovery was slow and required a stay in the ICU. One month after the initial intervention, a follow-up CT reported several mediastinal and retroperitoneal lymphadenopathies. In addition, in the previous tumor bed, a focal hepatic lesion was found in segment VIII, suggestive of metastasis, along with ascites and radiological signs of carcinomatosis. After an unfavorable evolution, the patient died 2 months after surgery.

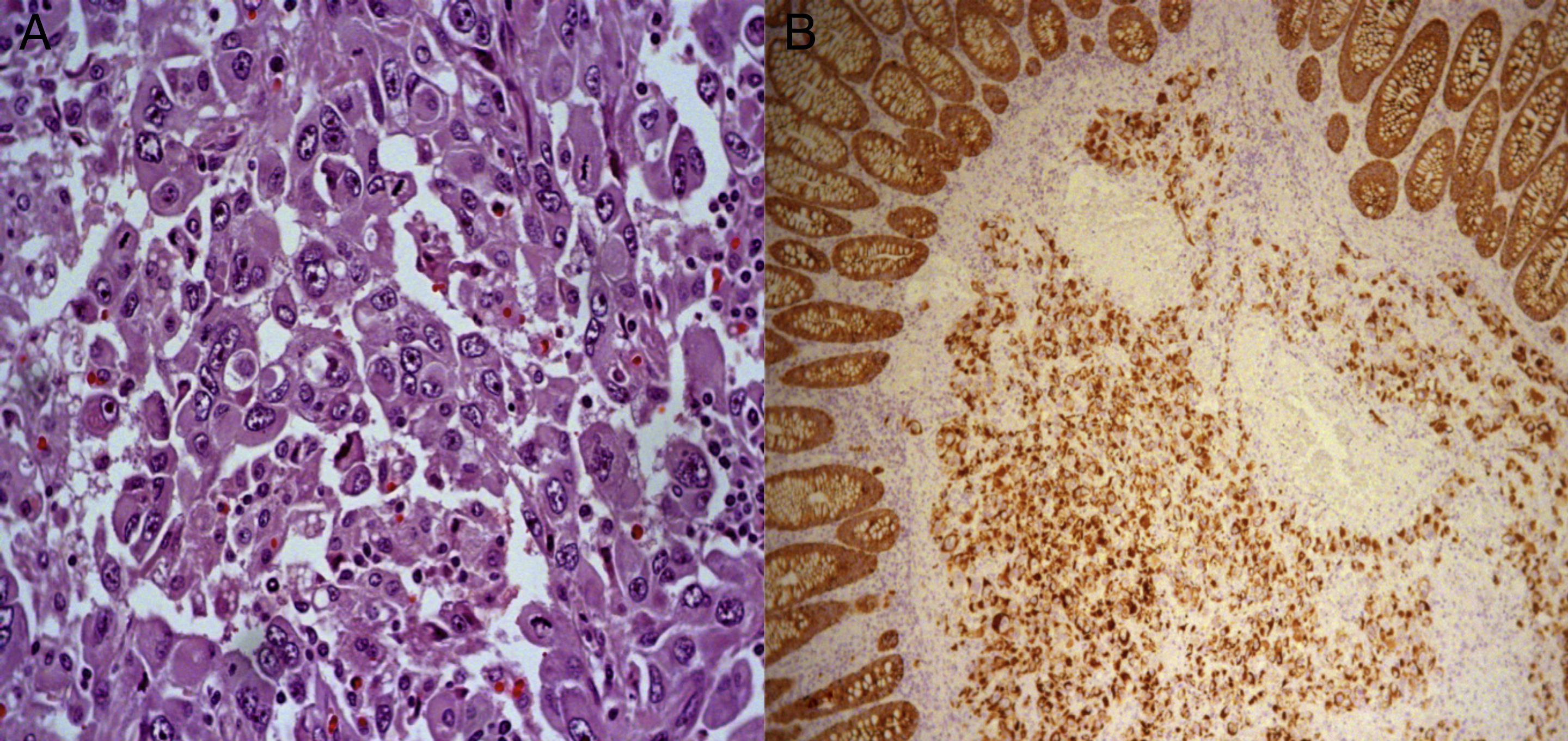
 Histology: hematoxylin–eosin stain (400×). Solid sheet of cells with extensive eosinophilic cytoplasm, with large vesicular, eccentric nuclei that give the cells a rhabdoid appearance. (B) Histology: cytokeratin cocktail (100×); positive neoplastic cells with cytokeratin. Note the internal control of the normal colonic epithelium.")
(A) Histology: hematoxylin–eosin stain (400×). Solid sheet of cells with extensive eosinophilic cytoplasm, with large vesicular, eccentric nuclei that give the cells a rhabdoid appearance. (B) Histology: cytokeratin cocktail (100×); positive neoplastic cells with cytokeratin. Note the internal control of the normal colonic epithelium.
Malignant rhabdoid tumors seem to preferentially settle in the kidneys, although they have also been reported in the brain, liver, skin, soft tissue, genital-urinary tract and gastrointestinal tract. Extrarenal malignant rhabdoid tumors present the same histological and immunohistochemical characteristics, aggressivity and poor prognosis as the renal forms. There is some controversy, however, about their histogenesis and denomination based on the fact that they could be considered either a phenotypic variation of the tumor depending on the organ of origin or an independent clinical-pathological entity. EMRT in pediatric patients are a well-defined clinical-pathological entity with morphologic, phenotypic and genetic findings (absence of nuclear expression of INI1 in tumor cells and specific genetic alterations in most cases, such as SMARCB1 gene inactivation). In contrast, most studies coincide in the opinion that rhabdoid tumors in adults form part of a phenotypic spectrum due to the sarcomatoid differentiation of tumor cells. Thus, their currently-accepted denomination is “poorly differentiated carcinoma with rhabdoid phenotype”.3
In adults, malignant rhabdoid tumors are a heterogeneous group of neoplasms with a common phenotypic expression (carcinomas, melanomas, sarcomas, histiocytes and lymphomas), which could be labeled malignant poorly differentiated carcinomas with rhabdoid phenotype according to whether the originating organ of the neoplastic tissue able to be identified. This occurred in our case with the initial biopsy obtained by colonoscopy, where it was questioned whether the origin was colonic. The rhabdoid phenotype is defined by the presence of pleomorphic cells with large, eccentric nuclei, prominent nucleoli, abundant and eosinophilic cytoplasm, paranuclear inclusions of intermediate filaments and abundant mitotic figures. Characteristic immunohistochemistry findings include the co-expression of cytokeratin and vimentin.5,6
In the literature, there are 12 published descriptions of EMRT in the colon.4 They are usually large tumors, most frequently located in the proximal colon and affecting patients with a mean age at diagnosis of 70, with no differences between sexes.2,5,7,8 They present a very aggressive clinical course with frequent metastasis at diagnosis; mean survival is 6 months, regardless of the tumor stage.2,5–8 Six cases have been classified as pure rhabdoid tumors, as in our case, while in the other 6 the rhabdoid cells coexisted with areas of adenocarcinoma. This supports the theory that they originate from the sarcomatoid differentiation of tumor cells.4
EMRT of the colon present limited response to treatment with radiotherapy and chemotherapy, so the treatment of choice is surgery.6–8 Given the aggressive nature of these tumors, however, in most cases it is merely a palliative treatment. In this context, the preoperative diagnosis of a rhabdoid tumor is key in order to plan treatment and avoid unnecessary surgery.9
In conclusion, the identification of the rhabdoid phenotype in the gastrointestinal tract is important as it is associated with a poor prognosis and a lack of response to conventional therapies.10
Please cite this article as: Romera Barba E, Sánchez Pérez A, Duque Pérez C, García Marcilla JA, Vázquez Rojas JL. Tumor rabdoide maligno de colon: a propósito de un caso. Cir Esp. 2014;92:638–640.
Presented as a poster at the 17th National Meeting of the Spanish Society of Coloproctology in Palma de Mallorca from May 8 to 10, 2013.

Cirugía Española (English Edition) sigue las recomendaciones para la preparación, presentación y publicación de trabajos académicos en revistas biomédicas