




El tumor sólido pseudopapilar de páncreas en un tumor epitelial extremadamente raro con bajo potencial de malignidad. Supone menos del 1-2% de todos los tumores pancreáticos exocrinos1. Fue descrito por primera vez en 19592, y desde entonces ha sido denominado de distintas formas: tumor papilar de páncreas o tumor de Frantz, neoplasia epitelial papilar sólida y quística o neoplasia quística papilar. Desde 1996 se le llama tumor sólido pseudopapilar de páncreas3. Es más frecuente en mujeres jóvenes o asiáticas de raza negra, entre 20 y 40 años de edad, aunque existen casos aislados en niños y en hombres.
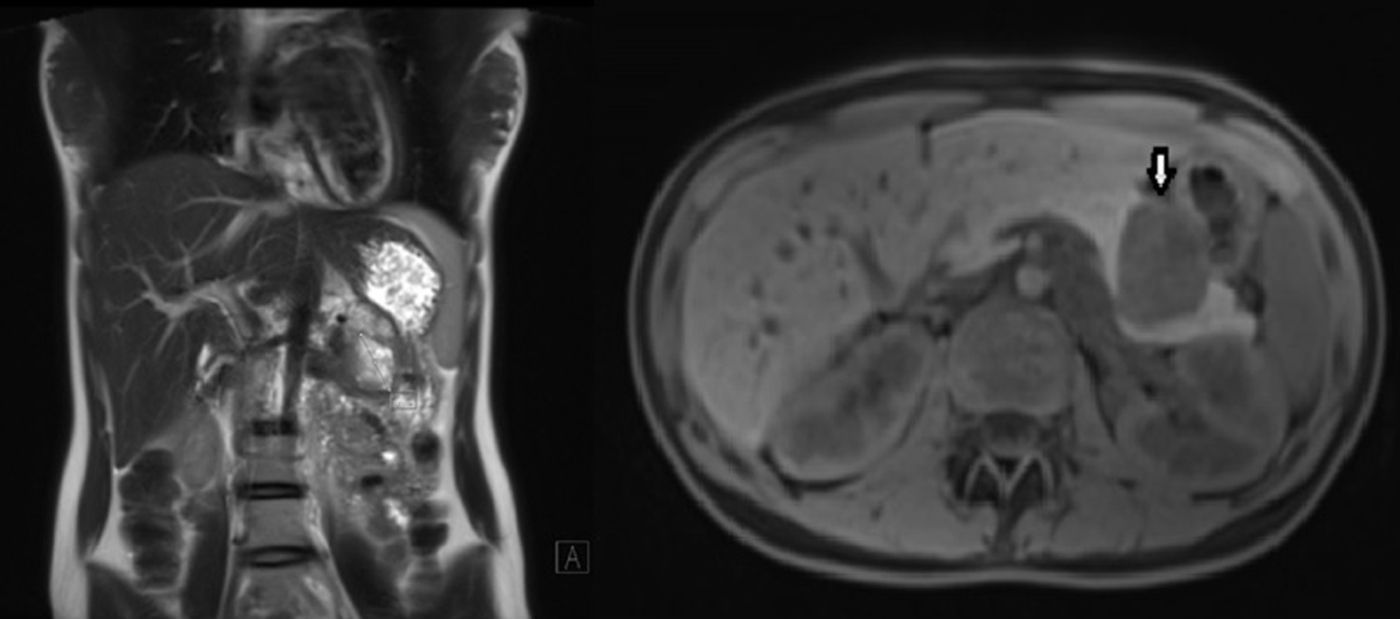
Presentamos el caso de una mujer de 17 años que consultó por dolor abdominal epigástrico y sensación de plenitud precoz de varios meses de evolución, sin otra sintomatología. Se realizó gastroscopia, que evidenció una compresión gástrica extrínseca a nivel del cuerpo gástrico, así como TAC abdominal y RMN (fig. 1), en las que se apreciaba una masa sólida retroperitoneal dependiente del cuerpo del páncreas de 5cm de diámetro. La ecoendoscopia mostró que se trataba de una lesión sólida hipervascular en el cuerpo-cola del páncreas. Se realizó una PAAF que indicó el diagnóstico de neoplasia sólida pseudopapilar de páncreas. La analítica, incluyendo los marcadores tumorales, se encontraba dentro de la normalidad.

Ante la sospecha diagnóstica se realizó una pancreatectomía corporocaudal laparoscópica con preservación esplénica y de los vasos esplénicos (técnica de Mallet-Guy laparoscópica)4 sin incidencias. El postoperatorio transcurrió con normalidad. Fue dada de alta al sexto día postoperatorio.
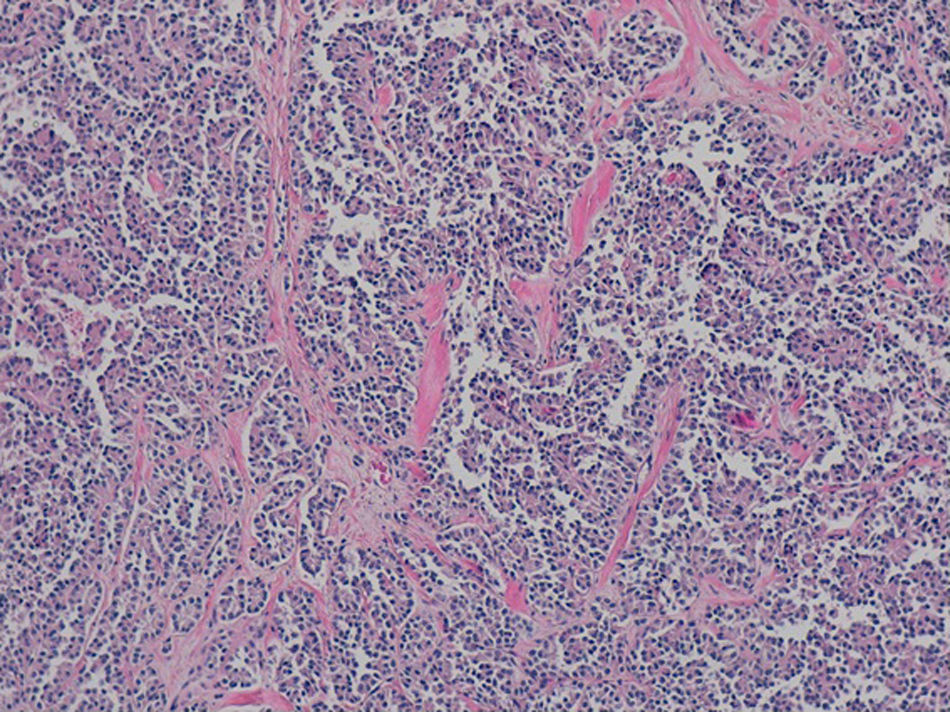
El análisis anatomopatológico definitivo confirmó el diagnóstico de neoplasia pseudopapilar de páncreas, sin invasión vascular ni perineural (fig. 2). El estudio inmunohistoquímico fue CD56, CD10 y beta-catenina fuertemente positivo. Progesterona y sinaptofisina con positividad focal, y citoqueratina AE1-AE3 y cromogranina negativos.
, y con presencia de glóbulos hialinos.")
El tumor sólido pseudopapilar de páncreas es una neoplasia pancreática muy poco frecuente de etiología desconocida que afecta principalmente a mujeres jóvenes en la segunda y tercera décadas de la vida. Se desconoce la etiología. Se ha propuesto un origen ductal epitelial, neuroendocrino, una célula primordial pluripotencial e incluso origen extrapancreático de origen genital5. El pronóstico es favorable aun en presencia de metástasis a distancia, habiéndose descrito supervivencias superiores a los 10 años incluso en presencia de metástasis hepáticas o peritoneales6. Las manifestaciones clínicas no son específicas, y se relacionan con el tamaño tumoral, incluyendo habitualmente dolor abdominal, sensación de plenitud o la presencia de masa abdominal7.
Los análisis de laboratorio suelen ser normales y la localización más frecuente es la cola del páncreas, seguida por el cuerpo8. Habitualmente el diagnóstico se realiza mediante pruebas de imagen (ecografía, TC y RMN) que muestran una masa bien circunscrita, encapsulada, heterogénea (sólido-quística), con calcificaciones ocasionales y áreas necróticas9. El diagnóstico diferencial se plantea con el cistoadenoma, cistoadenocarcinoma, neoplasias quísticas mucinosas, pancreatoblastomas, teratomas y tumores neuroendocrinos pancreáticos como lesiones hipervasculares más frecuentes. El diagnóstico de sospecha se presenta ante una lesión pancreática sólido-quística hipervascular en mujer joven, y, en caso de duda, la PAAF con ecoendoscopia puede confirmar el diagnóstico preoperatorio10,11. Para el diagnóstico diferencial con los tumores neuroendocrinos, que en su mayoría presentan receptores de somatostatina, podría realizarse el OctreoScan®, ya que la neoplasia sólida pseudopapilar carece de este tipo de receptores.
El diagnóstico de certeza se realiza mediante biopsia y el tratamiento recomendado es la resección quirúrgica. El 85% de los casos se encuentran limitados al páncreas en el momento del diagnóstico, habiendo metastatizado el resto en el momento del diagnóstico.
Las localizaciones más frecuentes de las metástasis son el hígado, ganglios regionales, el mesenterio, el epiplón y el peritoneo.
El tratamiento de elección es la cirugía, aunque, cuando se encuentra localizado, no se recomienda la realización de linfadenectomía. Frente a la presencia de metástasis o invasión local, la cirugía continúa siendo el tratamiento de elección. En el análisis anatomopatológico es típica la presencia de áreas sólidas alternando con zonas peudopapilares, si bien se ha descrito recientemente el aumento de la expresión nuclear y citoplasmática de la E-cadherina y la beta-catenina como marcadores específicos12.
La incidencia de neoplasia sólida pseudopapilar maligna o carcinoma sólido pseudopapilar es del 15%. Determinadas características histológicas se han asociado con un comportamiento agresivo, como el alto índice mitótico, atipias nucleares, necrosis extensa, áreas sarcomatoides y relacionadas con la expresión de Ki-6713. En este sentido, el Ki-67 se ha propuesto como indicador de potencial maligno, de forma que un índice bajo (inferior al 5%) indica un crecimiento tumoral lento y mejor pronóstico14,15. El papel de la quimioterapia o radioterapia adyuvante no está claro, aunque generalmente está reservado a casos irresecables.
Sin embargo, aunque la resección quirúrgica es generalmente curativa, se recomienda un seguimiento para diagnosticar recurrencias locales y metástasis a distancia. La supervivencia global a los 5 años es del 95%7,16,17.

