




La mayoría de los cánceres de recto son adenocarcinomas, pero existe un pequeño porcentaje de tumores de otras estirpes histológicas, como neoplasias neuroendocrinas, sarcomas, linfomas y carcinomas de células escamosas, que tienen unas características y tratamientos diferentes. Hemos efectuado una revisión de estos raros tumores del recto desde un punto de vista clínico y quirúrgico.
Most rectal neoplasms are adenocarcinomas, but there is a small percentage of tumors which are of other histological cell lines such as neuroendocrine tumors, sarcomas, lymphomas and squamous cell carcinomas, which have special characteristics and different treatments. We have reviewed these rare tumors of the rectum from a clinical and surgical point of view.
El cáncer de recto típico es el adenocarcinoma. No obstante, hay otros tipos de tumores que son mucho más raros, como las neoplasias neuroendocrinas, linfomas, sarcomas y carcinomas de células escamosas, que también pueden localizarse en el recto1–4. La incidencia de cada uno de estos tumores es difícil de precisar. Según los datos de los años 2005-2009 de la National Cancer Institute's Surveillance Epidemiology and End Results (SEER)5, de 183.000 cánceres colorrectales, entre los que no se incluía a los linfomas, 94,3% eran adenocarcinomas, 1,7% otros carcinomas, 3,3% tumores carcinoides, 0,5% carcinomas epidermoides, 0,1% sarcomas y 0,1% otros tipos.
Todos estos tumores presentan unas características muy diferentes a las de los adenocarcinomas, que hacen que su tratamiento y pronóstico sean muy distintos (tabla 1). Además, ha habido recientes modificaciones en el diagnóstico y tratamiento de alguno de ellos. Todo ello provoca que se generen dudas y controversias en su manejo clínico y se recomienda que sean tratados por un equipo multidisciplinar en el que se incluyan cirujanos, oncólogos, patólogos y radiólogos6–8.
Particularidades de cada tipo de tumor
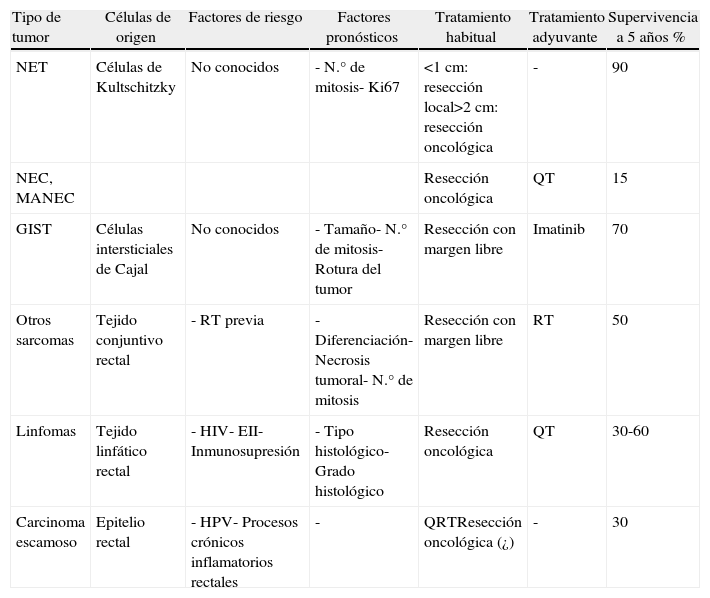
| Tipo de tumor | Células de origen | Factores de riesgo | Factores pronósticos | Tratamiento habitual | Tratamiento adyuvante | Supervivencia a 5 años % |
| NET | Células de Kultschitzky | No conocidos | - N.° de mitosis- Ki67 | <1cm: resección local>2cm: resección oncológica | - | 90 |
| NEC, MANEC | Resección oncológica | QT | 15 | |||
| GIST | Células intersticiales de Cajal | No conocidos | - Tamaño- N.° de mitosis- Rotura del tumor | Resección con margen libre | Imatinib | 70 |
| Otros sarcomas | Tejido conjuntivo rectal | - RT previa | - Diferenciación- Necrosis tumoral- N.° de mitosis | Resección con margen libre | RT | 50 |
| Linfomas | Tejido linfático rectal | - HIV- EII- Inmunosupresión | - Tipo histológico- Grado histológico | Resección oncológica | QT | 30-60 |
| Carcinoma escamoso | Epitelio rectal | - HPV- Procesos crónicos inflamatorios rectales | - | QRTResección oncológica (¿) | - | 30 |
EII: enfermedad inflamatoria intestinal; GIST: tumor del estroma gastrointestinal; HIV: virus de la inmunodeficiencia humana; HPV: virus del papiloma humano; MANEC: carcinoma mixto adenoneuroendocrino; NEC: carcinoma neuroendocrino; NET: tumor neuroendocrino; QRT: quimiorradioterapia; QT: quimioterapia; RT: radioterapia.
El objetivo de este artículo es el realizar una revisión sobre el manejo clínico y quirúrgico de estas neoplasias de recto poco frecuentes.
Se ha realizado una revisión en la literatura utilizando PubMed, desde el año 1997 hasta el 2012, con las palabras clave descritas al inicio y referidas a la localización rectal.
Neoplasias neuroendocrinasLas neoplasias neuroendocrinas son epiteliales y presentan diferenciación neuroendocrina9. Pueden localizarse en diferentes órganos.
Se clasifican10,11 por su grado de diferenciación (bien o pobremente diferenciados) y su grado histológico (G1, G2 y G3), basándose en el número de mitosis y en el índice Ki679,12–14. De esta manera forman 3 grupos diferentes: tumor neuroendocrino (NET); carcinoma neuroendocrino (NEC) y carcinoma adenoneuroendocrino mixto (MANEC)7,12.
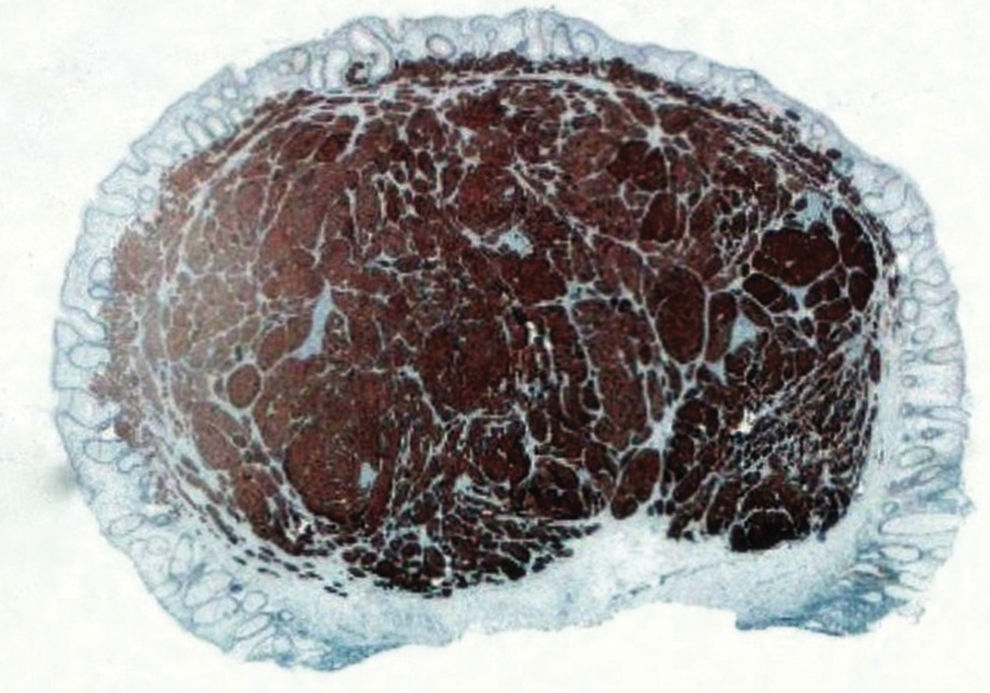
Tumores neuroendocrinosLos NET son unas neoplasias neuroendocrinas bien diferenciadas compuestas por células tumorales que expresan marcadores neuroendocrinos (cromagranina A, sinaptofisina) (fig. 1) y hormonas12. La atipia celular y su actividad proliferativa son bajas. Por definición, son tumores de grado G1 o G2. Esta categoría incluye los clasificados previamente como «tumor carcinoide», denominación actualmente criticada y que ya no se incluye en las clasificaciones de los NET digestivos, pero sigue utilizándose ampliamente. La localización rectal supone el 18% de todos los NET y el 27% de los del tubo digestivo12,15.

La incidencia anual de los NET rectales, según la SEER5, es de 0,86 por 100.000, con un gran incremento en las últimas décadas7 y mayor incidencia en orientales12. La edad media es de 56 años16.
Los NET se presentan, habitualmente, como pequeñas lesiones polipoides o nódulos submucosos4. El 45% miden 10mm o menos, mientras que solo el 17% miden más de 20 mm17. Con frecuencia son asintomáticos12,18 o se acompañan de síntomas leves como el sangrado, tenesmo o molestias17,18.
El 49% de los NET afectan únicamente a la mucosa y submucosa, el 24% infiltran la muscular propia y el 15% se extienden hasta la grasa perirrectal17. El 75-85% están localizados en la pared rectal12. El tamaño del tumor y la invasión linfovascular son factores de riesgo para afectación ganglionar16,17,19,20.
Las metástasis hepáticas son más frecuentes conforme se incrementa el tamaño del tumor16. Estas están presentes en el 1,7% de los NET de ≤ 1cm, en el 15% de los de 1-2cm y en el 50% de los de > 2 cm20.
La mayor parte de los NET se diagnostican endoscópicamente12. La ecografía endorrectal parece el método mejor para valorar el tamaño y profundidad de invasión de estos tumores16,18,20,21. En los NET de menos de 1cm y sin factores de riesgo no hacen falta más estudios. La RM y la TC estarán indicadas en los de mayor tamaño para estudiar la pelvis y descartar metástasis hepáticas12. El octreoescan parece tener una alta sensibilidad pero es poco utilizado20 y se emplea principalmente ante la sospecha de enfermedad metastásica12,18.
Menos del 1% de los NET colorrectales producen serotonina u otras hormonas, por lo que no se recomienda el análisis rutinario de serotonina ni de 5-HIAA18. La cromogranina A puede estar elevada y es de utilidad como marcador tumoral en el seguimiento de pacientes operados de estadio II y III o en enfermedad metastásica7,12,18.
El tamaño del tumor predice su comportamiento y el tipo de tratamiento a realizar12,16,17. También hay que tener en cuenta otros factores, como la invasión de la muscular propia, invasión linfovascular, atipia e índice mitótico12,20,22. El tratamiento de un NET localizado es la resección completa12–14.
Los NET de menos de 1cm pueden ser tratados mediante resección local12,18,19,21, ya que presentan menos del 3% de riesgo de metástasis ganglionares12. Previamente debe haberse descartado infiltración de la muscular propia mediante ecografía23. La resección puede realizarse con endoscopia estándar (fig. 1) o de doble canal22–27, mediante cirugía transanal17,21, o incluso mediante ligadura con bandas28.
El tratamiento de los NET de entre 1 y 2cm de tamaño no está claro, ya que tendrán metástasis ganglionares entre el 10 y el 15%12,18. La resección local estará indicada en los que se descarte afectación de la muscular propia y de los ganglios mediante ecografía18,21 y tengan bajo índice mitótico12. Si se reconoce atipia y alto índice mitótico, debería considerarse una cirugía radical12.
Los tumores de más de 2cm de tamaño tienen un riesgo de entre el 60 y el 80% de tener metástasis ganglionares12,18. Los NET de más de 2cm, o con invasión de la muscular propia, o con afectación ganglionar, deberán ser tratados con una resección anterior de recto o con una amputación abdominoperineal, según la distancia al margen anal12,18. No hay evidencia para tratamiento adyuvante tras la cirugía12.
Debe proponerse cirugía curativa a los pacientes con metástasis hepáticas operables, ya que se obtiene una supervivencia a los 5 años del 60-80%29. No se recomienda tratamiento adyuvante posterior29. Puede realizarse trasplante hepático en casos seleccionados, en los que no es posible la extirpación quirúrgica29.
En la enfermedad metastásica se ha utilizado octreótido long acting release (LAR) e interferón-α7,18,21,29. Recientemente se ha empleado radioterapia con péptidos análogos de la somatostatina marcados con radionúclidos, con respuestas del 30% en pacientes con tumores que expresan receptores de somatostatina7,18. Raramente se indica quimioterapia en NET G1 o G2. Cuando se usa por progresión de la enfermedad, se utiliza con mayor frecuencia estreptozotocina en combinación con 5-fluorouracilo±doxorrubicina, pero la respuesta es menor del 25%7,12.
La supervivencia a 5 años es del 91% en la enfermedad localizada, 49% en enfermedad regional y del 32% en enfermedad metastásica15.
El seguimiento no es necesario en los NET de < 1cm, sin otros datos de mal pronóstico13. En el resto, en el seguimiento se emplea la ecografía endorrectal, rectoscopia, RM o TC y cromogranina A13. Continuará hasta el 10.° año12.
Carcinomas neuroendocrinos y carcinomas mixtos adenoneuroendocrinosLos NEC son unas neoplasias malignas de alto grado, pobremente diferenciadas, de células tumorales que expresan marcadores neuroendocrinos (cromogranina A, sinaptofisina) y tienen una marcada atipia celular, frecuentes necrosis y alta actividad proliferativa12,30. Los NEC y los MANEC son tumores G3 por definición. Existen 2 categorías de NEC: de célula pequeña y de célula grande. Por sus características histológicas se comportan de una forma mucho más agresiva que los NET31,32.
La incidencia anual de estos carcinomas colorrectales es de 2 casos por 1.000.000 de habitantes2.
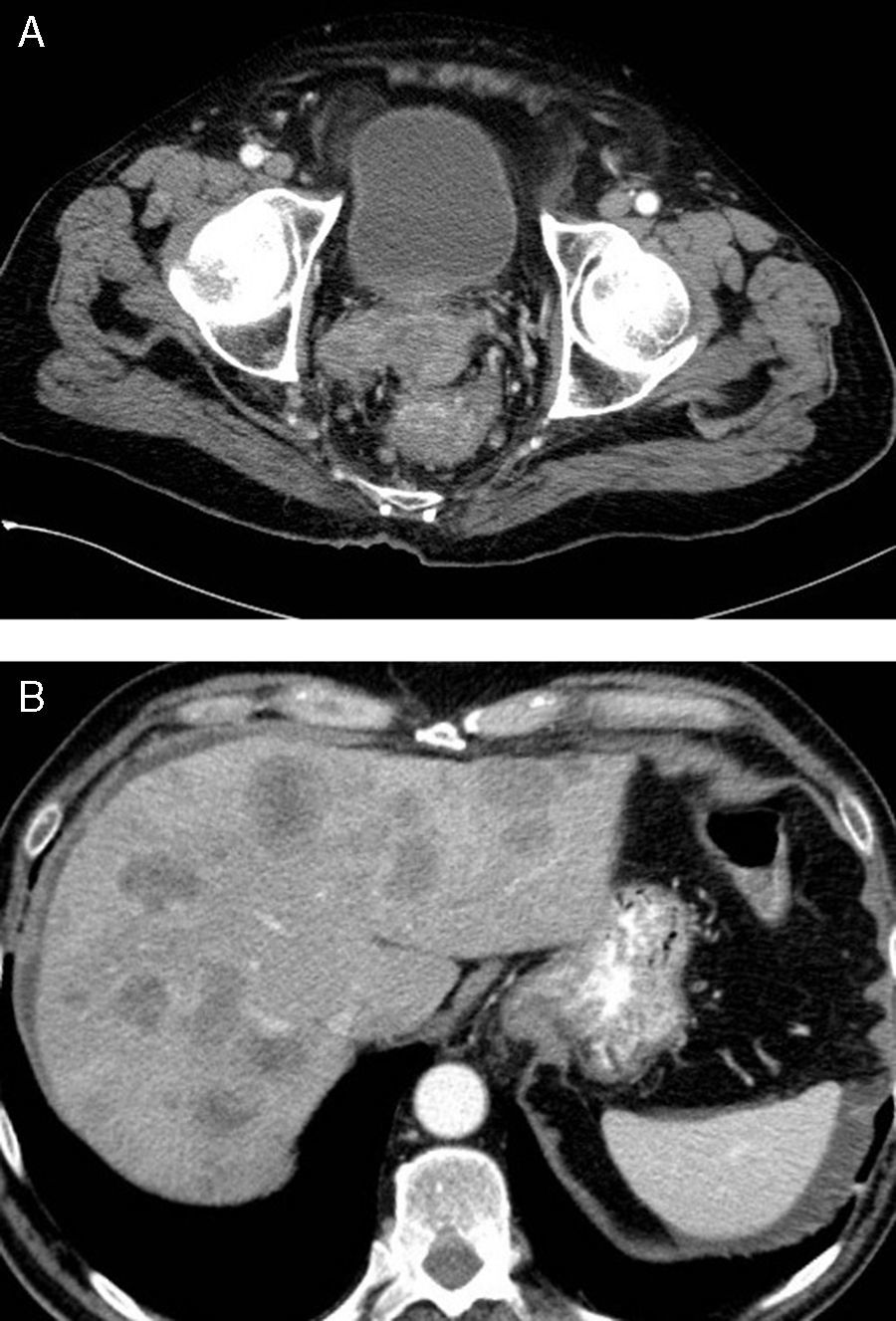
La sintomatología es similar a la de los adenocarcinomas de recto, pero se diferencian de estos en que en el momento del diagnóstico muchos de ellos ya tienen metástasis (fig. 2 a y b) y tienen peor pronóstico32. La supervivencia media es de 11 meses31,33.
 Carcinoma neuroendocrino rectal localmente avanzado y con metástasis hepáticas al inicio.")
En los NEC, la cromogranina A suele ser negativa, pero puede utilizarse la enolasa neuronal específica como marcador15.
No hay un tratamiento estandarizado30. El tratamiento habitual es quirúrgico: amputación o resección anterior según la localización, con exéresis mesorrectal30. No obstante, parece que la cirugía sola es curativa en pocas ocasiones por lo que se recomienda quimioterapia adyuvante en la mayoría de los casos30. Se emplea la misma quimioterapia que se utiliza en los carcinomas neuroendocrinos pulmonares, una combinación de cisplatino o carboplatino y etopósido7,29,30. La radioterapia puede estar indicada en casos de riesgo de recidiva local30. Se han descrito casos de buena evolución con quimiorradioterapia sola, sin cirugía34.
SarcomasHasta la década de los 90, la mayor parte de los tumores mesenquimales intestinales eran leiomiomas o leiomiosarcomas, pero con el desarrollo de técnicas inmunohistoquímicas se vio que la mayoría de estos tumores pertenecían a un grupo diferente, los gastrointestinal stromal tumor (GIST)35,36.
Tumor del estroma gastrointestinalLos GIST se originan en las células intersticiales de Cajal, que son unos marcapasos intestinales. Se caracterizan por tener unos marcadores específicos, como CD117 (c-KIT) en más del 95% y CD34 en el 70%, que los diferencian de los leiomiomas y leiomiosarcomas37. Se estima una incidencia anual de 1,5 por 100.0006. Las localizaciones habituales son la gástrica y la del intestino delgado, mientras que la rectal alcanza el 10% del total35.
Son tumores submucosos, mayoritariamente de entre 4 y 15cm. Pueden presentar una necrosis central y ulcerarse a la luz rectal. Los síntomas más frecuentes son el tenesmo y la rectorragia38.
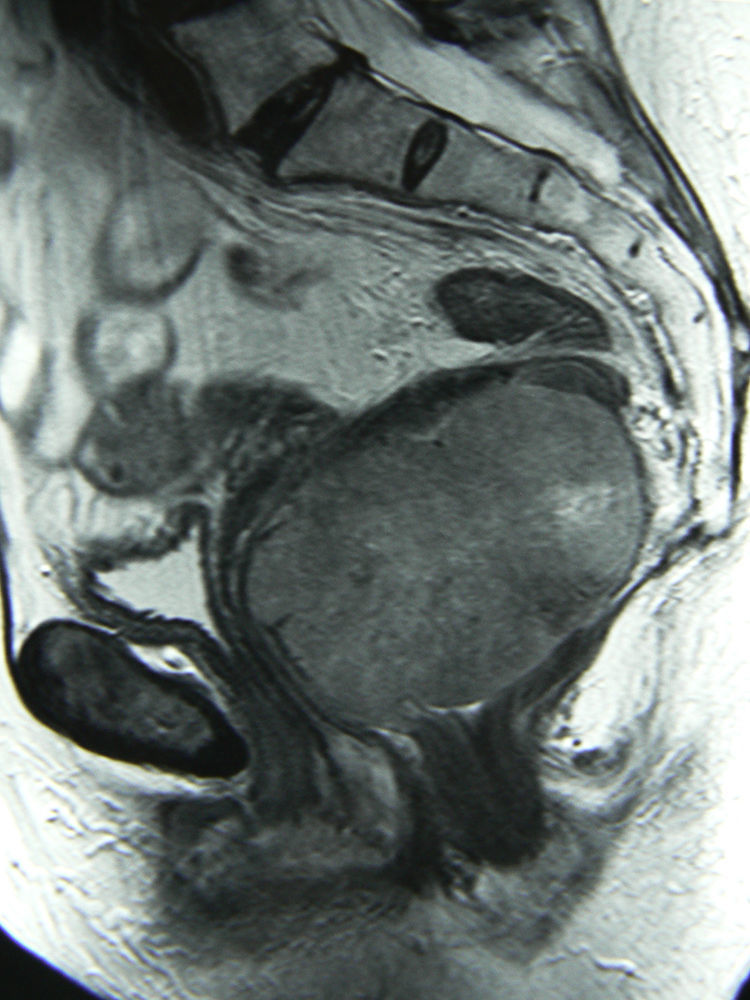
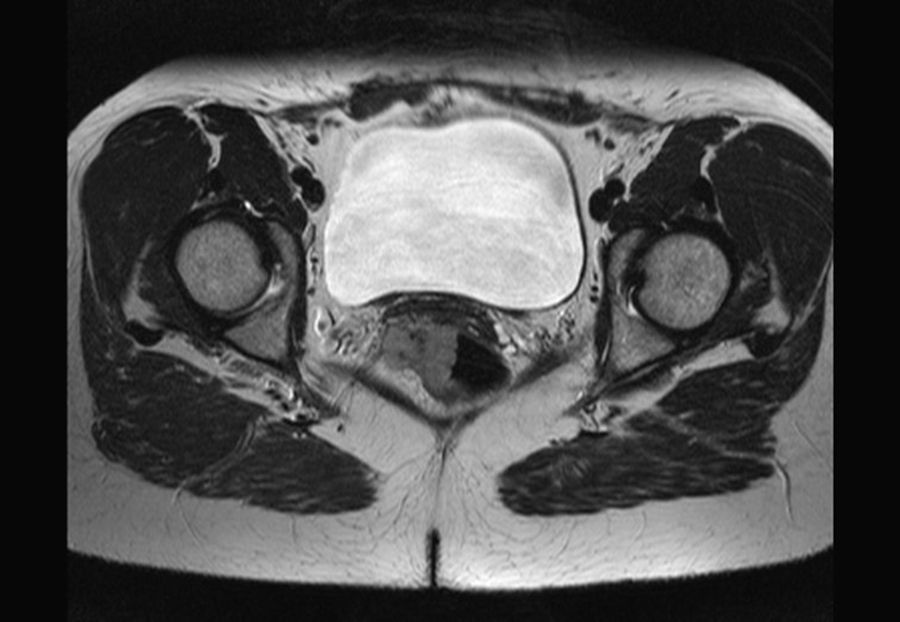
El diagnóstico se realiza por TC, RM y ecografía endorrectal. El GIST rectal se visualiza como una masa excéntrica con márgenes tumorales bien definidos (fig. 3), que puede tener áreas de hemorragia o necrosis39,40. La RM parece ser la exploración preferida en la localización rectal41. Mediante la ecografía se confirma que el tumor se origina en la pared muscular y no en la mucosa40. El diagnóstico histológico puede ser difícil, por no afectar a la mucosa38. En el caso de requerir cirugías amplias o de riesgo, o en caso de dudas diagnósticas con procesos que precisen otro tratamiento, se recomienda la biopsia ecoendoscópica, que tiene una tasa de éxito del 80-90%42.

El tratamiento estándar del GIST localizado es la resección quirúrgica con márgenes libres6,36,43,44, sin precisar de lifadenectomías si los ganglios son clínicamente negativos, ya que las metástasis ganglionares son poco frecuentes6,37. Estos tumores tienen una pseudocápsula y son friables, debiéndose manipular con cuidado para evitar su rotura37,42,43 ya que si se produce, empeora el pronóstico45.
En el recto, el tamaño y la localización del GIST determinan el tipo de cirugía. Los de pequeño tamaño pueden tratarse mediante una resección local. No debe haber células tumorales en el margen de resección6. Esta resección se puede realizar mediante cirugía abdominal o transanal46. No queda claro si tumores < 1cm, asintomáticos, deberían extirparse o seguir un control estricto, y resecarse solo si aumentan de tamaño6,42. Los tumores grandes, generalmente mayores de 5cm, son tratados habitualmente por resección anterior, sin necesidad de escisión mesorrectal, o mediante amputación abdominoperineal38,44,47,48. Se han descrito casos de tumores grandes extirpados por vía transacra49,50, o transvaginal51.
Los factores pronósticos más importantes son el tamaño del tumor y el número de mitosis y su localización6,36,43,52,53. La localización rectal tiene peor pronóstico que la gástrica6,45. Además, la rotura del tumor es un factor adverso45.
Es un tumor resistente a la quimioterapia, pero sensible a un inhibidor de los receptores tirosincinasa, el imatinib, que ha demostrado un importante beneficio clínico en pacientes con enfermedad avanzada o con recidiva37 y, también, que es eficaz para aumentar la supervivencia en pacientes de riesgo, mediante un tratamiento adyuvante54–56.
La supervivencia libre de enfermedad a los 5, 10 y 15 años es, respectivamente, del 70, 63 y 60%45. La adyuvancia está indicada en los pacientes con riesgo de recurrencia6,43. En los GIST rectales se recomienda en los de > 5cm con cualquier número de mitosis/50 campos, de cualquier tamaño con>5 mitosis/50 campos, o con rotura45,57. La dosis recomendada es de 400mg/día durante 3 años54,58.
Ante la buena respuesta de los GIST al imatinib, se está empleando este como tratamiento neoadyuvante para conseguir resecar tumores inicialmente irresecables y para evitar la amputación abdominoperineal en tumores distales grandes37,42,59–62. Tras la neoadyuvancia disminuye el tamaño tumoral y aumenta la resecabilidad60,62-64, incluso puede producirse una respuesta completa62,63,65. Estas indicaciones no están avaladas por estudios aleatorizados y se basan en series cortas o casos aislados. Recientemente, en una serie multicéntrica44, comprobaron que el tratamiento neoadyuvante disminuye el tamaño del tumor y aumenta la resecabilidad, pero no evita la cirugía mutilante, concluyendo, además, que la cirugía sigue siendo el tratamiento de elección en el GIST primario resecable. Antes de la neoadyuvancia debe haber una confirmación histológica66. La duración óptima del tratamiento preoperatorio es desconocida63,66,67. Para unos, la máxima respuesta tumoral se logra tras 3-6 meses de tratamiento40,68, mientras que, para otros, parece razonable seguir 6-12 meses63. El tratamiento debería mantenerse hasta llegar a la máxima respuesta, definida por la no mejoría entre 2 CT o RM63,66,67. El empleo del PET puede predecir la respuesta al tratamiento a las 2 semanas de haberse iniciado, ya que se manifiestan antes los resultados funcionales que los morfológicos66.
Para pacientes con tumor metastásico inoperable el tratamiento estándar es imatinib40,41,68. El tratamiento debe continuarse indefinidamente, ya que su interrupción se acompaña generalmente de una rápida progresión tumoral6,68. En caso de progresión durante el tratamiento con imatinib, se pueden emplear otras sustancias como el sunitinib, en segunda línea, o el regorafenib, en tercera línea6.
No hay datos para recomendar una pauta de seguimiento a los pacientes operados por un GIST localizado, pero parece lógico que se realice en función del riesgo6. La mayoría de las recidivas se producen en los primeros 5 años, siendo rara la recidiva tras 10 años45.
Otros sarcomasLos sarcomas de tejidos blandos y viscerales, excluyendo los GIST, tienen una incidencia anual estimada de 4-5 por 100.0008. Los sarcomas rectales son muy poco frecuentes, ya que los digestivos representan el 2,6% del total de sarcomas y, dentro de estos, solo el 15% son colorrectales69. Hay numerosos subtipos histológicos ya que se clasifican según las células de origen de este tejido. El más frecuente, en el recto, es el leimiosarcoma69. El grado histológico de malignidad (G1, G2 o G3) viene determinado por 3 parámetros: la diferenciación, la necrosis tumoral y el número de mitosis8,70.
No hay una etiología clara de estos tumores, pero ya es conocido el aumento de riesgo de aparición de sarcomas tras radiaciones ionizantes. Aparecen, generalmente a los 7-10 años de la radioterapia71. Se han descrito leiomiosarcomas y angiosarcomas tras radioterapia pélvica72–75. Por otra parte, el sarcoma de Kaposi se presenta asociado al sida76.
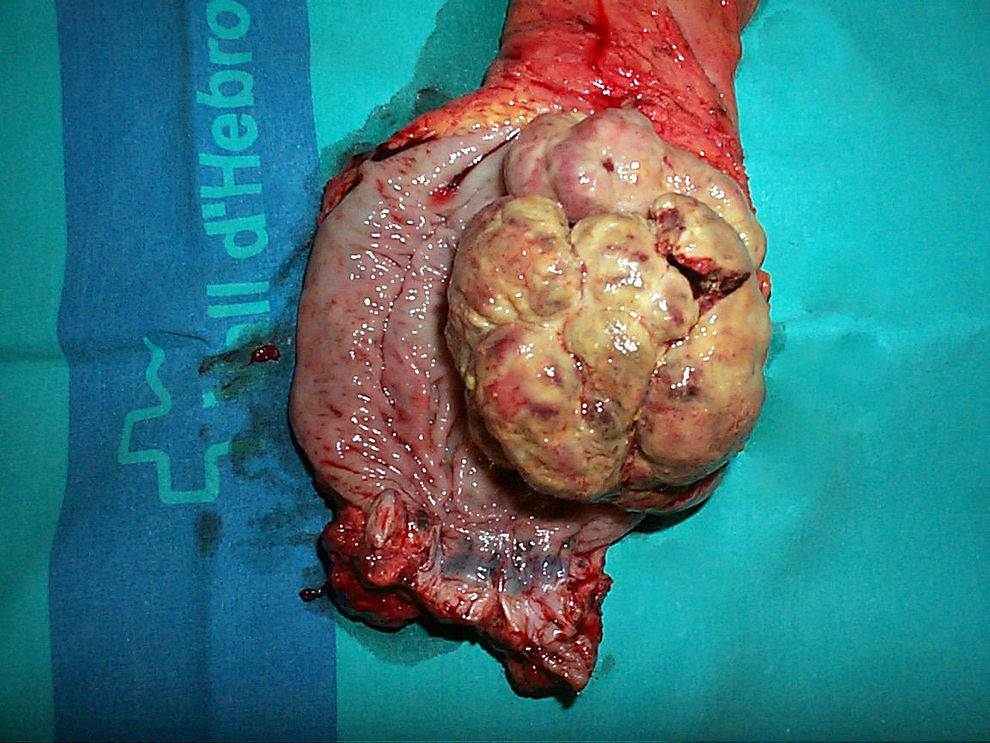
Los leiomiosarcomas tienen diferente origen que los GIST: derivan de las células de la muscularis mucosa o de la muscular propia36,47. En el estudio inmunohistoquímico presentan positividad a desmina y a actina de músculo liso y negatividad a CD117 y CD34, que los diferencian de los GIST47,70. Con frecuencia se presentan como lesiones polipoides de entre 2 y 5 cm47 (fig. 4). Acostumbran a ser tumores bien diferenciados y con alta actividad mitótica, pero parece que su pronóstico puede ser mejor que los GIST con similar número de mitosis47.

El grado histológico, el tamaño y la invasión de órganos adyacentes determinan su pronóstico69,70.
La resección quirúrgica con márgenes libres es el tratamiento de elección69. Las metástasis ganglionares son poco frecuentes8. No queda claro si una resección local es suficiente para tumores pequeños de bajo grado69. La indicación de resección anterior o amputación abominoperineal se hará en función del tamaño y localización del tumor. La radioterapia puede tener resultados similares a los que se obtienen en el tratamiento de los sarcomas de las extremidades y se recomienda en tumores mayores de 5cm y alto grado77.
Las recidivas se presentan como metástasis hepáticas, pulmonares o locales en la pelvis. Según sus características podrá realizarse resección quirúrgica, ablación, radioterapia o quimioterapia8. Al no existir un marcador específico, el seguimiento debe realizarse con pruebas de imagen8.
LinfomasEl linfoma gastrointestinal es raro, pero es la localización extraganglionar más frecuente de linfomas no Hodgkin (LNH). La afectación colorrectal es más rara que la gástrica y la del intestino delgado. Se considera que el linfoma es primario cuando no hay afectación sistémica, es decir, en ausencia de adenopatías periféricas, sin afectación linfática mediastínica, con estudio hematológico en sangre periférica y biopsia de médula ósea normales, con adenopatías localizadas únicamente en la proximidad de la lesión y sin afectación hepática ni esplénica78,79.
El linfoma rectal secundario es un proceso generalizado con afectación rectal por metástasis ganglionares. La diferenciación entre linfoma primario y secundario es importante ya que el tratamiento y pronóstico son distintos. El tratamiento del linfoma secundario es quimioterápico y la supervivencia a 5 años es del 15%1.
En el recto, pueden presentarse todos los subtipos histológicos de linfomas, pero la mayor parte de los linfomas primarios son LNH de células B79–81, con sus diferentes variantes: de células B grandes, de células del manto, folicular, de Burkitt y tipo mucosa-associated lymphoid tissue (MALT)78,82,83. La proporción de los diferentes subtipos varía según áreas geográficas79. Los LNH de células T son más frecuentes en Oriente que en Occidente79,80.
Diferentes factores se han implicado en la génesis de los linfomas gastrointestinales, generalmente asociados a inmunosupresión, como la infección HIV, enfermedad inflamatoria intestinal, trasplante de órganos o tratamiento con corticoides79,80. También se han relacionado con agentes infecciosos, como el H. pylori, y otros más80.
La edad media al diagnóstico es de 55 años78,79. Los síntomas más frecuentes son dolor abdominal, pérdida de peso, cambio en el ritmo intestinal y rectorragia79,81.
Aunque en el colon se presenta como una lesión polipoide, ulcerada o no, masa estenosante, poliposis segmentaria, o nodularidad mucosa79,80,83, en el recto es más típica una masa homogénea, por engrosamiento concéntrico parietal, con estenosis de la luz39,84. La existencia de adenopatías grandes y numerosas son sospechosas de linfoma79. La biopsia endoscópica con estudio inmunohistoquímico dará el diagnóstico, aunque en muchos casos, el diagnóstico exacto preoperatorio puede ser difícil81,85–87.
Debido al escaso número de pacientes y a los varios subtipos histológicos, no hay un tratamiento estandarizado de los linfomas colorrectales79,88. Se puede utilizar una combinación de cirugía y quimioterapia, reservando la radioterapia a algunos casos82,83.
La resección quirúrgica oncológica es el tratamiento habitual para los linfomas localizados78,79,81–83 ya que ofrece la posibilidad de cura sin tratamiento adyuvante y previene de las complicaciones como el sangrado, obstrucción o perforación82,85.
La quimioterapia como tratamiento inicial se reserva habitualmente a pacientes con tumor localmente avanzado81 o enfermedad diseminada83.
La quimioterapia adyuvante, tras la cirugía, se recomienda en linfomas agresivos o estadios avanzados79,85. El régimen quimioterápico más empleado es el CHOP (ciclofosfamida, doxorrubicina, vincristina y prednisona)78,82,83,85,89. Añadiendo un anticuerpo monoclonal anti CD-20, el rituximab, a este esquema clásico (R-CHOP), mejoran los resultados79.
La radioterapia adyuvante podría tener algún papel en el control locorregional después de una resección incompleta82,83 o en casos de tumores quimiorresistentes79.
Los raros linfomas MALT rectales han sido tratados de muy diversas formas. En algunos Helicobacter pylori positivos se ha utilizado con éxito la antibioterapia sola90,91, en otros se ha empleado la radioterapia, la quimioterapia o la cirugía92.
La supervivencia a 5 años de los linfomas colorrectales está entre 25-57%, que es peor que la de los gástricos y la del intestino delgado82.
Carcinoma escamosoEl carcinoma escamoso es un tumor poco frecuente, que habitualmente se localiza en esófago y en ano. La afectación rectal es muy rara. En muchos casos los presuntos carcinomas escamosos rectales son, en realidad, una extensión de un carcinoma de ano93. Ocasionalmente tienen un patrón histológico mixto y se denominan carcinoma adenoescamoso94.
La media de edad es de 57 años y es algo más frecuente en mujeres que en hombres93,95. No hay factores de riesgo claros, pero se ha encontrado asociado a enfermedades inflamatorias de recto, infecciones del virus del papiloma humano (HPV) y adenocarcinoma colorrectal95.
Los síntomas son similares a los del adenocarcinoma de recto, siendo el más frecuente la rectorragia93,95. Los hallazgos endoscópicos pueden mostrar desde una formación polipoidea hasta un tumor ulcerado y estenosante95. La biopsia dará el diagnóstico. Ocasionalmente puede existir dificultad para distinguirlo de un tumor pobremente diferenciado, pero la inmunohistoquímica puede caracterizar la lesión95. Las citoqueratinas más útiles son CAM 5.2, AE1/AE3 y 34B1295.
Para la estadificación se emplea la RM (fig. 5), TC y ecografía endorrectal. El antígeno de carcinoma de células escamosas es un marcador tumoral que está elevado en algunos de los pacientes. No se utiliza para el diagnóstico, pero sí puede utilizarse para monitorizar la respuesta o la progresión95,96.

Clásicamente, el tratamiento habitual era el quirúrgico, seguido en algunos casos de quimio- o radioterapia adyuvante97,98. En la última década, ante los buenos resultados de la quimiorradioterapia en el carcinoma escamoso de ano, se ha cuestionado este enfoque clásico99 y, a pesar de no estar establecido un tratamiento estándar93, hay una tendencia a modificar las pautas, convirtiéndose la quimiorradioterapia en el tratamiento inicial del carcinoma escamoso de recto, y reservándose la cirugía a la persistencia tumoral tras el tratamiento93,96,100,101. Se utilizan las mismas pautas que han demostrado su eficacia en el carcinoma escamoso de ano95. Se emplea una combinación de mitomicina-C con 5-fluorouracilo y radioterapia, con una dosis mínima de 45-50 Gy102–104. La valoración de la respuesta a la quimiorradioterapia se realiza a las 6-8 semanas de finalizado el tratamiento mediante rectoscopia con biopsia, RM o PET93,99–101. Si hay respuesta clínica y radiológica completa se realizarán controles periódicos y, en caso de persistencia tumoral, se podrá reevaluar a las 4-6 semanas como se recomienda en el cáncer anal104, con cirugía de rescate si se precisa, mediante resección anterior o amputación, en función de las características del tumor y del paciente97.
Las series publicadas en el que el tratamiento inicial fue la quimiorradioterapia son escasas y con pocos casos, pero entre el 66 y el 100% mostraron respuesta completa y no precisaron de cirugía posterior93,96,99–101. Tras respuesta completa se deberán seguir controles clínicos, con realización de biopsias rectales y controles radiológicos, que se irán espaciando progresivamente99.
La supervivencia a 5 años es del 50% en el estadio II y desciende al 33% cuando hay afectación ganglionar95.
ConclusionesLa rareza de estos tumores y su heterogéneo origen, tratamiento y pronóstico hacen que los especialistas clínicos puedan tener dificultades en el manejo de estos pacientes. Se recomienda un enfoque multidisciplinar en el que se incluyan patólogos, radiólogos, oncólogos, radioterapeutas y cirujanos.
Conflicto de interesesLos autores declaran no tener ningún conflicto de intereses.

