




El tumor fibromixoide osificante es un tumor de partes blandas, englobado dentro de los tumores mixoides, grupo heterogéneo de lesiones que tienen en común la producción de matriz extracelular rica en mucopolisacáridos. Es poco frecuente (menos de 300 casos descritos) y presenta un crecimiento lento con gran capacidad para la recidiva local y la metástasis.
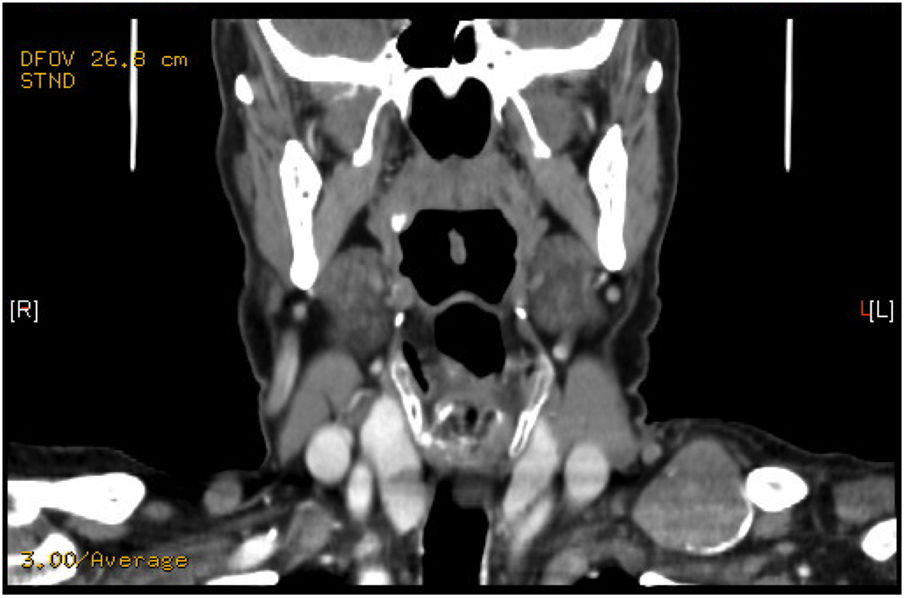
Presentamos el caso de un varón de 74 años que presenta tumoración supraclavicular de 4cm de diámetro, de reciente aparición, no dolorosa, dura y parcialmente móvil a la exploración. En ecografía aparece un nódulo sólido-quístico de 32mm en contacto con la clavícula. La PAAF informa de neoplasia de probable estirpe neuroendocrina. El estudio hormonal resulta dentro de la normalidad. La TAC observa una lesión supraclavicular de 3cm en el triángulo cervical posterior izquierdo (fig. 1) y el SPECT-TAC aprecia un depósito focal de moderada a elevada densidad en la fosa supraclavicular izquierda. Se decide intervención quirúrgica apreciándose masa de 4cm de diámetro en el espacio supraclavicular extendida por detrás la clavícula de consistencia dura con áreas calcificadas. Se realiza exéresis quirúrgica. El paciente evoluciona de forma favorable, siendo dado de alta en 24h.

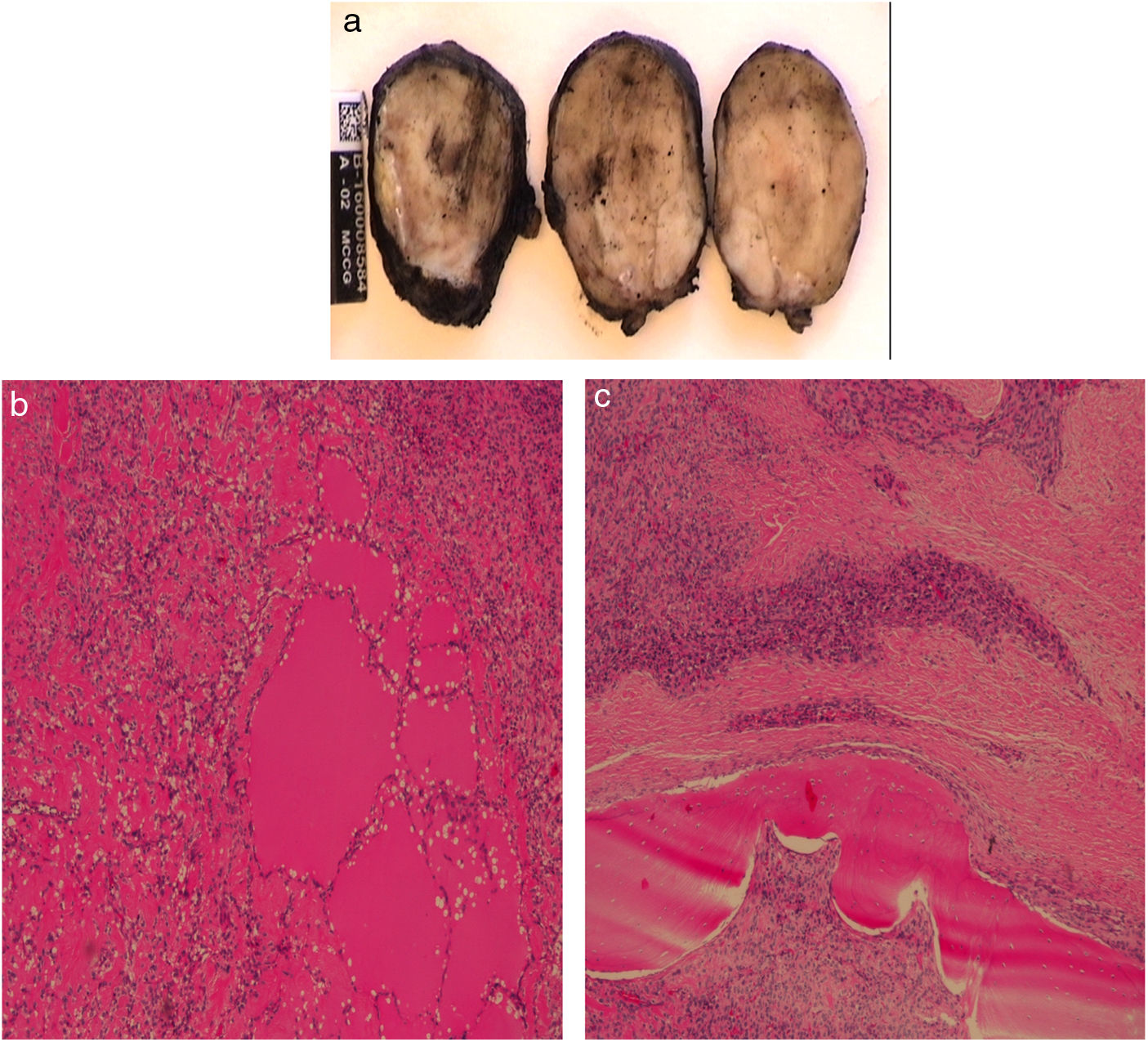
La anatomía patológica informa de una formación ovoidea de 4×4×2,5cm, correspondiente a una tumoración mesenquimal multinodular seudoencapsulada (fig. 2A). La lesión es hipercelular con áreas colagenizadas, hábito epitelioide, moderado citoplasma eosinófilo y núcleos irregulares. Presenta 5 mitosis/50CGA que en algunas zonas alcanzan hasta 2-3/10CGA y áreas de osificación en forma de anillo concéntrico. Se identifican algunos focos de aspecto mixoide compatible con tumor fibromixoide calcificante maligno (figs. 2B y C). En el comité de tumores se decide administrar radioterapia adyuvante permaneciendo tras 2 años de seguimiento, libre de enfermedad.
 Se observa una formación ovoidea correspondiente a una tumoración mesenquimal multinodular seudoencapsulada. B y C) Se identifican algunos focos de aspecto mixoide compatible con tumor fibromixoide osificante maligno.")
Los tumores fibromixoides osificantes ocurren en adultos de cualquier edad, sobre todo en varones en torno a los 50 años1 en el subcutáneo de tejidos blandos o músculos esqueléticos, el 70% aparecen en extremidades (proximales más que distales), existiendo casos descritos en tronco, cabeza, cuello, mediastino, retroperitoneo o mama. Suelen ser nódulos pequeños con un tamaño promedio de 4-5cm1,2, bien circunscritos, indoloros y profundos, adheridos a los tendones subyacentes, fascia o músculos esqueléticos. Suelen presentar un crecimiento de meses a años de duración (media de 4 años)1.
Descrito por primera vez por Enzinger et al.3,4 en 1989, basándose en 59 casos del Instituto Patológico de las Fuerzas Armadas en 25 años, como una masa nodular bien circunscrita en el tejido subcutáneo o los músculos, denominándolo tumor fibromixoide osificante de partes blandas.
La gran mayoría son histológicamente benignos con un pronóstico favorable5. Sin embargo, hay casos con características citoarquitecturales atípicas y rasgos histológicos de malignidad (alta celularidad, atipia nuclear y actividad mitótica elevada >2/50CGA), que presentan mayor recurrencia y capacidad para metastatizar (60% de malignos)4,6. Es característica la existencia de cordones de células blandas pequeñas con matriz rica en mucopolisacáridos y frecuente hialinización parietal3. Pueden expresar tejido óseo en el 60-90% de los casos y necrosis hasta en el 20%7.
No existe una clasificación de riesgo aceptada universalmente. Folpe y Weiss2 describen 70 casos, y los divide en 3 tipos. Los tumores «típicos» son aquellos con celularidad y grado nuclear bajos, falta de necrosis o invasión vascular, con actividad mitótica <2/50CGA. Los «atípicos» son aquellos que se diferencian de los típicos, pero sin criterios de malignidad. Los «malignos» presentan una tasa de recurrencia y metástasis del 60%. En su serie publicada, 8 pacientes de 70 casos tuvieron metástasis a distancia2,7 (principalmente pulmón).
En la inmunohistoquímica expresan la proteína S100, sobre todo en los tumores «típicos»5,8. Antonescu et al.8 observaron la expresión S100 en el 60% de los casos y la positividad de EAAT4, MUC4, NFP y CD56.7,16 CD10 en tumores «atípicos». Graham et al.9 describen un mosaico característico de patrones de pérdida de INI1/SMARCB1. En un pequeño número de casos el gen INI1 está ausente, correspondiendo con la pérdida de expresión nuclear7. Los estudios con FISH han demostrado un reordenamiento recurrente del gen PHF1 (situado en cromosoma 6p21) entre el 49-79% de los casos.
Radiológicamente se comporta como una masa nodular de tejido blando con un anillo de osificación incompleto. La TAC muestra una «concha de hueso» (Bone-shell) periférica entre el 60-70% de los casos1,2,5. Los exámenes con tecnecio muestran la presencia de formación intratumoral de hueso maduro1. En la RMN el tumor suele ser isointenso a los músculos en T1 y mostrar intensidad intermedia-alta en T210.
El tratamiento habitual es la resección completa de la lesión con márgenes libres, lo que suele ser curativo en la mayoría de los casos4. Sin embargo, dado que la variante maligna tiene riesgo de recurrencia y metástasis se añade radioterapia adyuvante del lecho tumoral.
El tumor fibromixoide osificante maligno es muy raro, con una alta tasa de recurrencia y metástasis, siendo muy importante su diagnóstico mediante métodos histológicos, inmunohistoquímicos y FISH. Es necesario un mejor conocimiento de estos tumores con el fin de evitar errores en el diagnóstico y contribuir a su correcto manejo para aumentar la supervivencia.

