




Familial hypercholesterolaemia (FH) is the autosomal dominant genetic disorder most frequently associated with premature cardiovascular disease (CVD).
Materials and methodsA retrospective, observational study was conducted to determine the clinical characteristics, analytical parameters and cardiovascular risk factors of 133 patients with a genetically confirmed diagnosis of FH on follow-up in the Lipid Clinic of Alava.
ResultsCVD was observed in 8.30% of the patients (ischaemic heart disease in 100% of the cases). The LDL concentration goal was achieved in 40.6% (45.50% in primary prevention and 27.30% in secondary prevention). The large majority (81.80%) of patients with coronary heart disease (CHD) were male. The odds ratio (OR) of males having CHD compared to females is 4.97 (1.03–23.93, p=0.03). The OR of developing CHD in patients with a family history of premature CVD is 6.86 (1.32–35.67, p=p.02). A statistically significant association was found between smoking and the risk of CVD (p=0.005), and also between having diabetes and the risk of CVD (p=0.0001). If the treatment with statins begins at older than 40 years, the OR of suffering CHD is 6.40 (1.53–26.5) (p=0.009). The mean time from diagnosis to the cardiovascular event in the group of ex-smokers is 10.80±5.80 years, and in the non-smoking group it is 17.50±2.50 years (p=0.011).
ConclusionsIn our reference population with FH, it was found that there was an increased risk of suffering a cardiovascular event in male patients, with a family history of premature CVD, diabetics, and in those in whom lipid lowering treatment was started after 40 years of age.
La hipercolesterolemia familiar (HF) es el trastorno genético autosómico dominante más frecuentemente asociado a enfermedad cardiovascular (ECV) prematura.
Material y métodosEstudio observacional, retrospectivo, para determinar las características clínicas, los parámetros analíticos y los factores de riesgo cardiovascular de 133 pacientes con diagnóstico genético confirmado de HF en seguimiento en la Unidad de Lípidos de Álava.
ResultadosEl 8,30% de los pacientes ha presentado ECV (en el 100% de los casos cardiopatía isquémica [CI]). El 40,60% alcanza el objetivo de cLDL: el 45,50% en prevención primaria y el 27,30% en prevención secundaria. El 81,80% de los pacientes con CI son varones. El odds ratio (OR) de presentar CI en los varones frente a las mujeres es 4,97 (1,03–23,93; p=0,03). El OR de presentar CI en los pacientes con historia familiar de ECV prematura es 6,86 (1,32–35,67; p=0,02). Encontramos una asociación estadísticamente significativa entre fumar y el riesgo de ECV (p=0,005) y también entre tener diabetes y el riesgo de ECV (p=0,0001). Si el tratamiento con estatinas se inicia antes de los 40 años, el OR de presentar CI es 6,40 (1,53–26,50; p=0,009). El tiempo medio desde el diagnóstico hasta el evento en el grupo de exfumadores es 10,80±5,80 años y en el grupo de no fumadores es 17,50±2,50 años (p=0,01).
ConclusionesEn nuestra población de referencia con HF, encontramos un mayor riesgo de presentar un evento cardiovascular en los pacientes varones, con antecedentes familiares de ECV prematura, diabéticos y en los que se ha iniciado el tratamiento hipolipidemiante después de los 40 años de edad.
Familial hypercholesterolaemia (FH) causes elevated LDL cholesterol (LDL-C) concentrations from birth, and is consequently the autosomal dominant genetic disorder most commonly associated with premature cardiovascular disease (CVD). In the Caucasian population, estimates generally place the rate of heterozygous FH at 1/500 and homozygous FH at 1/1,000,000.1 However, the Copenhagen General Population Study found that the prevalence of confirmed or probable FH, defined as a score >5 according to the Dutch Lipid Clinic Network (DLCN) criteria, was 1/200.2 In Spain, the estimated prevalence of FH standardised by age and gender is 1/192 individuals for the heterozygous phenotype and 1/425,774 for the homozygous phenotype.3
In heterozygous patients, the mutated gene is the LDL receptor (LDLR) gene in over 90% of cases, the APOB gene in approximately 5% and the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene in approximately 1%.4 Patients with FH have a 3- to 13-fold higher risk of premature CVD than individuals without FH.2,5 In patients with heterozygous FH who do not receive lipid-lowering treatment, coronary disease typically develops before the age of 55 (males) or 60 (females), while homozygous patients will develop coronary disease before the age of 20 if they do not receive treatment.5
The SAFEHEART (Spanish Familial Hypercholesterolaemia) cohort study estimated that the prevalence of diabetes among patients with FH is 3.20%, 14.40% among those with hypertension (HTN) and 26.40% among those who are active smokers. Moreover, 9.40% of these patients have developed premature CVD.6
The aims of this study were to identify the clinical characteristics of patients with FH, determine the degree of lipid control and analyse the main cardiovascular risk factors (CVRF) among patients undergoing follow-up by the Lipid Clinic at Hospital Universitario Araba.
Materials and methodsStudy design and populationThis was a retrospective, observational study of 133 patients with a genetically confirmed diagnosis of FH undergoing follow-up by the Lipid Clinic at Hospital Universitario Araba. The study began including patients in 2001 (the year in which the Lipid Unit was accredited by the Sociedad Española de Arterioesclerosis [Spanish Atherosclerosis Society]) and continued until the last patients were included in June 2017. Both index cases and family cases were included. All patients were over the age of 18, except for two cases of recently diagnosed family members (aged 11 and 13, respectively).
Study variablesDemographic data were collected (age, gender, age at diagnosis of FH, age when lipid-lowering therapy was started, etc.), as well as physical examination data (weight, height, presence of corneal arcus and tendinous xanthomas, etc.) and analytical test data (total cholesterol [TC], LDL-C, HDL-C, TG, Lp[a], etc.). In the statistical analysis of the lipid profile data, the two patients aged under 18 and the patient with a diagnosis of homozygous FH were excluded to prevent extreme values from altering the mean of the rest of the data.
CVRF, such as diabetes, hypertension, smoking, family history of premature CVD, etc. were also analysed. Patients were diagnosed as having abnormal baseline blood glucose if their fasting blood glucose was ≥100mg/dl, diabetes if their baseline blood glucose was ≥126mg/dl or HbA1c ≥6.5% and prediabetes if their HbA1c was from 5.7% to 6.4%.
Last of all, we evaluated the type of lipid-lowering therapy used and adherence to it (estimated from the data from the electronic prescriptions on the number of packs of each drug collected in the previous six months), and any associated adverse reactions. The statins used were classified as high-potency (LDL-C reduction ≥50%), moderate-potency (LDL-C reduction from 30% to <50%) and low-potency (LDL-C reduction <30%). The follow-up target levels for LDL-C were defined according to the consensus document for the diagnosis and treatment of FH in Spain, taking into account age, associated CVRF and whether the patient was in primary or secondary prevention.7
Enzymatic methods were used to measure TC, TG, LDL-C and HDL-C levels. LDL-C levels were calculated using the Friedewald formula. Lipoprotein a (Lp[a]) levels were measured by a turbidimetric method (Quantia Lp(a)-Test, Abbott). The clinical diagnosis of FH was made based on the DLCN criteria; genetic testing is usually requested when the DLCN score is equal to or greater than 6 points. Genetic diagnosis was performed on blood samples (LIPOchip, Progenika®), with the relevant prior informed consent. A GE Logiq ultrasound system (GE Healthcare®) was used to perform the supra-aortic trunk (SAT) Doppler, with a high-frequency linear probe (7.5–15MHz). The plaque was defined as a focal structure that protruded more than 0.5mm into the artery lumen or by more than 50% of the intima-media thickness of an adjoining area or any intima-media thickness ≥1.5mm. A Microlife WatchBP Office ABI® dual digital blood pressure device was used to measure the ankle-brachial index (ABI); a value <0.9 was considered abnormal.
Statistical analysisQuantitative variables were analysed for normal distribution (Kolmogorov–Smirnov normality test). If the distribution was normal, the values are presented as mean±standard deviation (minimum–maximum); otherwise, they are expressed as median (P25–P75). Student's t-test was used for paired samples when distribution was normal and the Wilcoxon test otherwise.
Categorical variables are expressed as a percentage, comparing the proportions between groups using χ squared (χ2) or Fisher's exact test (if the sample sizes were small). The odds ratio (OR) was used to calculate any association between CVRF and CVD. The level of statistical significance was set at p<0.05. The time from diagnosis to the cardiovascular event was compared using Kaplan–Meier survival analysis.
For the management and statistical analysis of the data, the statistical program IBM SPSS Statistics 22® and Epidat 3.1® were used.
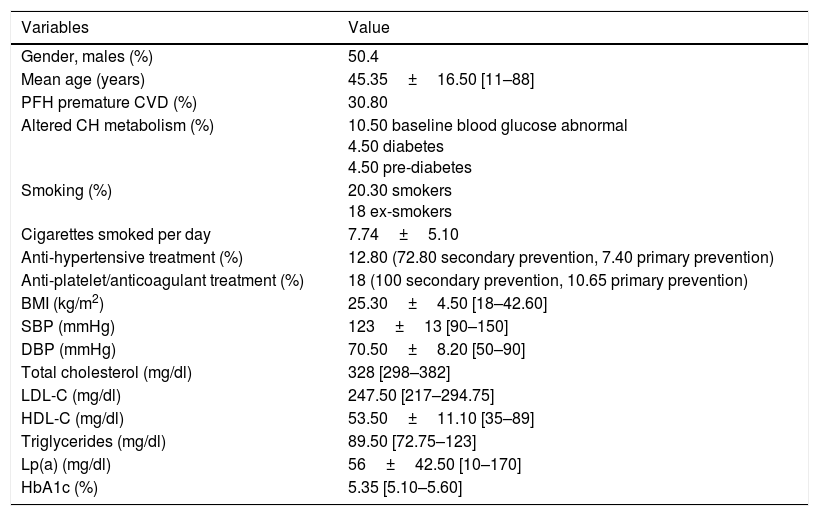
ResultsThe sample consisted of 133 patients with a confirmed genetic diagnosis of FH. The demographic characteristics (gender, age, mean age at diagnosis, average age when treatment was started), clinical characteristics (diabetes, smoking, blood pressure, BMI, anti-platelet treatment) and initial lipid values are shown in Table 1.
Sociodemographic and clinical characteristics and baseline lipid values.
| Variables | Value |
|---|---|
| Gender, males (%) | 50.4 |
| Mean age (years) | 45.35±16.50 [11–88] |
| PFH premature CVD (%) | 30.80 |
| Altered CH metabolism (%) | 10.50 baseline blood glucose abnormal 4.50 diabetes 4.50 pre-diabetes |
| Smoking (%) | 20.30 smokers 18 ex-smokers |
| Cigarettes smoked per day | 7.74±5.10 |
| Anti-hypertensive treatment (%) | 12.80 (72.80 secondary prevention, 7.40 primary prevention) |
| Anti-platelet/anticoagulant treatment (%) | 18 (100 secondary prevention, 10.65 primary prevention) |
| BMI (kg/m2) | 25.30±4.50 [18–42.60] |
| SBP (mmHg) | 123±13 [90–150] |
| DBP (mmHg) | 70.50±8.20 [50–90] |
| Total cholesterol (mg/dl) | 328 [298–382] |
| LDL-C (mg/dl) | 247.50 [217–294.75] |
| HDL-C (mg/dl) | 53.50±11.10 [35–89] |
| Triglycerides (mg/dl) | 89.50 [72.75–123] |
| Lp(a) (mg/dl) | 56±42.50 [10–170] |
| HbA1c (%) | 5.35 [5.10–5.60] |
BMI: body mass index; CH: carbohydrates; CVD: cardiovascular disease; DBP: diastolic blood pressure; PFH: previous family history; SBP: systolic blood pressure.
In terms of the type of genetic mutation, 78.50% were simple heterozygous mutations, 20.80% had a double mutation and one patient (0.80%) was homozygous. In 95.40% of cases, the mutated gene was found in LDLR. In 3.10% of patients, the mutation was located in the APOB gene and in 1.50% in the APOE gene. In our sample, the mean score according to the DLCN criteria was 11.70±3.80 (5–21) and 10.50% of our patients had a null allele mutation. The most common mutations in our sample were c.313+1G>C in combination with c.274C>G (amino acid change plus splicing, 17 patients); c.313+2dupT (splicing, 9 patients) and deletion of exon 5 (8 patients).
The TC at diagnosis without lipid-lowering treatment (328 [298–382] mg/dl) was significantly higher than the TC in the last year (180 [160–200] mg/dl, p=0.000). LDL-C at diagnosis (247.50 [217–294.75] mg/dl) was significantly higher than the mean LDL-C in the last year (115 [99–129.5] mg/dl, p=0.000). We found no statistically significant differences in the Lp(a) levels before and after the start of lipid-lowering treatment (p=0.31). In 14.30% of our patients, Lp(a) was undetectable. We also found no statistically significant differences in HbA1c levels before and after the start of lipid-lowering treatment (p=0.08).
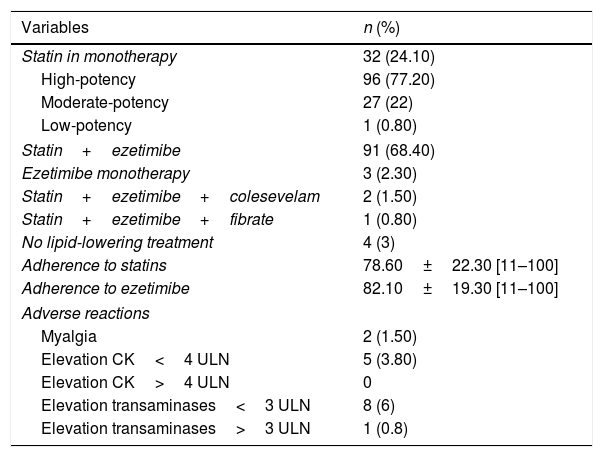
The characteristics of the lipid-lowering treatment are shown in Table 2. Interestingly, we found no statistically significant differences related to gender in adherence to either statins (p=0.80) or ezetimibe (p=0.58).
Lipid-lowering treatment.
| Variables | n (%) |
|---|---|
| Statin in monotherapy | 32 (24.10) |
| High-potency | 96 (77.20) |
| Moderate-potency | 27 (22) |
| Low-potency | 1 (0.80) |
| Statin+ezetimibe | 91 (68.40) |
| Ezetimibe monotherapy | 3 (2.30) |
| Statin+ezetimibe+colesevelam | 2 (1.50) |
| Statin+ezetimibe+fibrate | 1 (0.80) |
| No lipid-lowering treatment | 4 (3) |
| Adherence to statins | 78.60±22.30 [11–100] |
| Adherence to ezetimibe | 82.10±19.30 [11–100] |
| Adverse reactions | |
| Myalgia | 2 (1.50) |
| Elevation CK<4 ULN | 5 (3.80) |
| Elevation CK>4 ULN | 0 |
| Elevation transaminases<3 ULN | 8 (6) |
| Elevation transaminases>3 ULN | 1 (0.8) |
CK: creatine kinase; ULN: upper limit of normal.
Out of the total, 91.70% of the patients had not had any cardiovascular events (primary prevention) and 8.30% were on secondary prevention (in all cases because of coronary heart disease [CHD]); 40.60% achieved the target LDL-C (45.50% in primary prevention and 27.30% in secondary prevention). Of the patients with CHD, 81.80% were male; the OR of developing CHD in men compared to women was 4.97 (1.03–23.93, p=0.03). The OR of developing CHD in patients with a family history of premature CVD was 6.86 (1.32–35.67, p=0.02).
However, there was no statistically significant association between developing CVD and having an abnormal ABI (p=0.08) or having plaques show up in the SAT Doppler (p=0.59). Among patients with CHD, only 33.30% had an ABI<0.9; 44.40% were found to have atheromatous plaques in the SAT Doppler, although without haemodynamic repercussions, and none had vascular stenosis. The only patient who had stenosis <50% was in the group without CVD and none of our patients had vascular stenosis >50%.
We found a statistically significant association between smoking and the risk of CVD (p=0.005). The OR in former smokers (54% of patients with CHD) was 6.50 (1.66–25.5) and in smokers, it was 0.75 (0.08–7.02). Therefore, there is no linear relationship between exposure to tobacco and increased cardiovascular risk (p=0.55).
There is a statistically significant association between having diabetes and the risk of CVD (p=0.0001). The OR in patients with diabetes is 25.75 (3.90–169.80) and in patients with prediabetes, 12.90 (1.8–92.3). There is a linear relationship between the degree of alteration of carbohydrate metabolism and the risk of CVD (p=0.001). However, in our sample, there was no association between the risk of developing diabetes and the potency of the statin used (p=0.99).
There is a statistically significant association between the diagnosis of FH prior to generalisation of statin use (using 1995 as the cut-off point) and the risk of CHD (p=0.01). In fact, the risk of suffering a cardiovascular event is 5.80 (1.42–23.97) times higher if the patient was diagnosed with FH before the use of statins became generalised. There are no statistically significant differences between starting treatment with statins before or after the age of 18 and the risk of developing CHD (p=0.213). However, if the treatment with statins begins after the age of 40, the OR of developing CHD is 6.40 (1.53–26.50, p=0.01).
Statistically significant differences were found in the time from diagnosis of FH to the cardiovascular event based on exposure to tobacco (p=0.01). The mean time from diagnosis to the event in the ex-smoker group was 10.80±5.80 years and, in the non-smoker group, it was 17.50±2.50 years. However, we did not find statistically significant differences in the time to the cardiovascular event in terms of diabetes (p=0.51), gender (p=0.40), family history of premature CVD (p=0.13) or the potency of the statin used (p=0.42).
In the 6 patients on treatment with PCSK9 inhibitors (evolocumab), the mean TC was 154±46mg/dl (100–210) and the mean LDL-C was 77.30±32mg/dl (30–110). The mean reductions in TC and in LDL-C among patients on treatment with evolocumab were 40.50%±14.60% and 56.30%±17.20%, respectively.
DiscussionAssuming that FH affects 1/500 people5, on average, approximately 6% of patients in Spain are estimated to have been diagnosed. With a reference population of 324,126 according to Spanish National Institute of Statistics data from 2016, 20.50% of patients with FH in the province of Álava should therefore have been diagnosed. That means that the percentage of patients diagnosed in Álava is higher than the average for Spain. However, if we consider that the actual rate of FH is closer to 1/200, we would only have diagnosed 8.20% of these patients.2
Of our patients, 50.40% were male, the average age was 45.35 and 30.80% had a family history of premature CVD. The data are similar to those obtained in the SAFEHEART registry, with 45.2% males, an average age of 45.50 and a family history of premature CVD in 41.20% of cases.8 In our sample, 12.80% of patients were on antihypertensive treatment, 4.50% had diabetes and 20.30% were smokers. Concordant data were obtained in the SAFEHEART cohort, in which 14.20% of patients were hypertensive, 4.30% had type 2 diabetes and 25.60% were active smokers.8 Baseline levels of TC and LDL-C in patients with a definitive clinical diagnosis of FH in the Spanish Atherosclerosis Society registry are 340mg/dl and 260mg/dl, respectively, similar to the values found in this study (TC: 342.50mg/dl and LDL-C: 262.10mg/dl).9
In our study, 95.40% of the patients had a mutation of the LDLR gene, 3.10% in the APOB gene and 1.50% in the APOE gene. The data are compatible with previous studies which estimate that the mutation causing FH is LDLR in more than 90% of cases and the APOB gene in approximately 5%.4 In our sample, one family had a mutation in the APOE gene. Next-generation sequencing studies have shown that very occasionally FH is caused by dominant mutations in the APOE gene, which encodes apolipoprotein E, or in the STAP1 gene.10
In our study, 8.30% of the patients developed CHD, similar to the 11.80% of patients with atherosclerotic disease of the coronary arteries in the SAFEHEART registry.6 Of our patients, the 27.30% in secondary prevention achieved LDL-C<70mg/dl, similar to the data obtained in EUROASPIRE IV, with 80.50% of patients with LDL-C>70mg/dl.11 However, our lipid target data for patients in secondary prevention are better than the results obtained in the SAFEHEART registry, in which only 4.70% of patients achieved LDL-C<70mg/dl during follow-up.12
The risk of developing CHD in our study was almost five times higher in males than in females. In contrast, in the study conducted by Zamora et al., there was no statistically significant association between gender and risk of CHD in a population with the FH phenotype.3 However, an association between being male and increased risk of CHD had already been established by Hopkins et al.; in their study of 266 patients with heterozygous FH, they found the risk of CHD to be 5.64 times higher in males.13
Unlike the SAFEHEART registry, in which no association was found between a family history of premature CVD and a higher degree of cardiovascular risk, in our study, patients with a family history of premature CVD had an almost seven times greater risk of suffering an event.6 That is higher than the findings of deGoma et al., who reported an adjusted OR for all other CVRF of 1.84 in patients with a family history of premature CVD.14
We found a very strong association between diabetes and CVD in our study: the risk of CHD was 25 times higher in patients with diabetes. Our figures are much higher than those found in the SAFEHEART study, in which the OR for diabetes was 1.58.6 They are, however, in line with those obtained by De Sauvage Nolting et al., who demonstrated a risk of CVD 17.61 times higher in patients with diabetes.15
The main limitation of our study is that it was retrospective and observational and the reliability of our conclusions is not therefore comparable to that of a randomised clinical trial. However, it has allowed us to perform a descriptive analysis of patients with FH in our reference population and estimate which risk factors may be more directly associated with CVD. Nevertheless, the sample size is not comparable to the SAFEHEART study and the associations found with the different CVRF may therefore be exclusive to our reference population. In other words, it is possible that they cannot be fully extrapolated to the rest of the Spanish population with FH.
ConclusionsIn our reference population with FH, we found an increased risk of suffering a cardiovascular event in male patients, patients with a family history of premature CVD, patients with diabetes, and patients who did not start lipid-lowering therapy until they were over the age of 40.
To Luis Irigoyen at Hospital Clínico Universitario Lozano Blesa de Zaragoza, head of the Álava Lipid Clinic until 2014.
Please cite this article as: Pérez García L. Hipercolesterolemia familiar: experiencia en la Unidad de Lípidos de Álava. Clin Invest Arterioscler. 2018;30:224–229.