




New advances in the treatment of acute myocardial infarction involve novel signalling pathways and cellular progeny. In this sense, regeneration is a novel tool that would contribute to post-infarction physiological ventricular remodeling. More specifically, re-expression of the WT1 transcription factor in the myocardial wall by ischaemia and infarction would be related to the invasion of cells with the capacity for regeneration. This mechanism seems not to be sufficient to restore muscle cells and lost vessels entirely. Of particular interest, the presence of the heat-shock response protein 70 (Hsp70) and its interaction with the vitamin D receptor would modulate the expression of WT1 positively. In this context, it is proposed that the activation of vitamin D receptors associated with Hsp70 could favour physiological cardiac remodeling and reduce the progression to heart failure.
Nuevos avances en el tratamiento del infarto agudo de miocardio involucran inéditas vías de señalización y progenie celular. En este sentido, la regeneración es una herramienta novedosa que contribuiría al remodelado ventricular fisiológico postinfarto. Más específicamente, la reexpresión del factor de transcripción WT1 en la pared miocárdica por isquemia e infarto se relacionaría con la invasión de células con la capacidad de regeneración. Este mecanismo parece no resultar suficiente para restituir completamente las células musculares y vasos perdidos. De especial interés, la presencia de la proteína de respuesta a golpe de calor 70 (Hsp70) y su interacción con el receptor de vitamina D modularían positivamente la expresión de WT1. En este contexto, se propone que la activación de los receptores de vitamina D asociados a Hsp70 podría favorecer el remodelado cardiaco fisiológico y reducir la progresión a insuficiencia cardiaca.
Coronary heart disease was and continues to be a scourge of humanity. This is because myocardial cell death caused by deficient irrigation of the heart gives rise to premature death or the onset of heart failure associated with a reduction in the force of contraction or pathological cardiac remodeling. Of epidemiological interest, there is a close correlation between the current growing social demand to which humans are subjected and the greater predisposition to disease. Patients who suffer coronary heart disease over time experience the consequences of coronary events like heart failure and/or arrhythmia. This gives rise to various degrees of disability in the following years. Cardiovascular disease (CVD) is the leading cause of death worldwide. In 2013, it was responsible for 17.3 million deaths, 7.4 million of which were caused by coronary heart disease and 6.7 million by cerebrovascular accidents (CVA).1 Based on these data, it is estimated that this figure will rise to more than 23.6 million deaths by 2030. Specifically with regard to the 2013 figures, it is calculated that CVD caused 16 million deaths worldwide and was responsible for the loss of 293 million life years adjusted for disability, which represents around 31% of all deaths. The latest statistics obtained reveal that coronary heart disease is responsible for one in every seven deaths in the United States of America and kills more than 360,000 people every year. Together with CVA, it continues to be the leading cause of death.2
Post-myocardial infarction cardiac remodelingThe risk of death due to acute myocardial infarction (AMI) ranges from 6.5% to 8.5%.3 The mortality rate rises to 8.8% for ST-segment elevation AMI (STEMI), but falls to 5.1% for non-ST-segment elevation AMI (NSTEMI).4 This event can be associated with heart failure through the progression and extent of the myocardial damage. Despite the fact that the incidence of heart failure in AMI is very high, no study reflects the true extent of the problem due to recording difficulties and non-hospitalised infarctions. In these cases, the 10-year prognosis of patients with heart failure is poor and survival rates peak at just 10% when also associated with coronary heart disease.5
In the clinical course towards heart failure, the process of post-infarction ventricular remodeling is characterised by increased ventricular size and poor prognosis in patients in whom genetic, structural and biochemical abnormalities negatively affect the heart's long-term functional capacity and give rise to progressive heart failure.5
Infarct expansion is defined as the thinning of the wall in the infarcted region and dilation of the ventricular cavity. This expansion causes the shape of the heart to become more rounded and spherical. It is known that an increase in wall stress stimulates the replication of sarcomeres, with a preference for series replication. The interaction of certain factors, predominantly increased wall stress and inflammation, gives rise to an increase in the radius of the cavity/thickness of the wall ratio, which characterises chronic eccentric ventricular hypertrophy.6
Despite the conceptual prevalence that ventricular remodeling is associated with changes in the structure and size of the heart resulting from the progressive deterioration of ventricular function, there is still a lack of complete and compelling information concerning the responsible mechanisms. This assertion is supported by the findings of recent studies that include classical and other unpublished factors such as energy deficit, changes in the transport of calcium, adrenergic pathway abnormalities, changes in contractile proteins, apoptosis, inflammation, oxidative stress, metalloproteinase abnormalities,7 the regulation of the expression of organogenic genes like WT1 (factor Wilms’ tumour-1), vitamin D levels and heat shock proteins with a molecular weight of 70kDa (Hsp70).8–10
Of particular interest, these factors could be modulated by the use of drugs like angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), beta-blockers and statins, which have been proven effective in reducing the degree of remodeling and improving the long-term prognosis.11,12
The inflammatory activity caused by AMI affects remodeling and the normal progression of heart scarring. In this regard, it has been shown that increased post-AMI C-reactive protein (CRP) levels predict cardiac rupture and the onset of ventricular aneurysm over time, as well as an increased rate of cardiac death within one year of the occurrence of the event.13
Inflammation in response to myocardial necrosis is a physiological reaction that prepares for tissue healing through fibrosis (physiological-allostatic mechanism) but which may be excessive in the event of added factors like mechanical stress, autoimmunity, genetic background, neurohumoral activation, oxidative stress and ageing (allostatic overload),7 which ultimately gives rise to pathological remodeling. As such, myocardial damage and cardiac remodeling require the damaged structure and subsequent changes to be associated with a certain size and with a significant inflammatory response.6 The extent of the infarction should exceed 16–20% of the myocardial area of the left ventricle in order for remodeling to occur. In this regard, Armstrong studied a large group of patients with clear evidence of remodeling and characterised them into two subgroups; a larger group with dilated anterior myocardial infarction (61%) and a second group (33%) with inferior infarction.14
The accompanying inflammatory response, together with mechanical disorders (size of the infarction, wall stress) determine the change in configuration and structure of the heart, with subsequent pathological remodeling and abnormalities.
New developments in pathological cardiac remodeling associated with transcription factor WT1 and related factorsThe WT1 gene encodes a DNA-binding protein that acts as a transcriptional activator or repressor for more than 20 different genes by binding to their promoters. It is found in human chromosome 11p13 and measures 345kb. The mRNA of WT1 has three translation sites that give rise to three WT1 protein isoforms of different molecular weights: 62–64kDa, 52–54kDa and 36–38kDa. The main protein is the isoform weighing 52–54kDa. Its activity or function is controlled by phosphorylation of protein kinase A (PKA), which facilitates the translocation of WT1 from the nucleus to the cell cytosol. This process interferes with the transcriptional activities of WT1 that are vital for the development and maintenance of the heart, kidneys, mesothelium and gonads.15
With specific regard to the development of the heart, the pericardium supplies cardiogenic progenitor cells which, together with the epicardium and myocardium, direct lineage specification, determining both the growth of the myocardium as well as the formation of coronary vessels. The role of the adult pericardium is to offer a smooth surface for heart movement. However, in pathological conditions its mechanical function is impaired, a feature which has been studied in greater detail in recent years. In particular, recent findings highlight new functions associated with ischaemia and cardiac injury. The first evidence of epicardial involvement in cardiac regeneration emerged from studies in lower vertebrates.
The embryonic heart tube is formed by the fusion of heart fields that give rise to the left ventricle and parts of the atria. The progenitor cells of the proepicardium migrate to form the epicardium and contribute to the vasculature of the heart by moving from the surface to the endocardium. This event is perfectly modulated by WT1 expression, which remains quiescent after development. WT1 expression in the epicardium decreases after birth and remains low for the duration of adult life. In particular, WT1 endothelial expression in the adult heart is low and is primarily seen in certain heart capillaries and veins.16 In this regard, WT1 silencing in endothelial cells gives rise to a significant reduction in the formation of heart vessels and appropriate vessel networks.17 This underlines the importance of WT1 and the essential role it plays in cardiac endothelial cells.
Extremely relevant to this review is the very recent finding that ischaemia and cardiac necrosis cause WT1 overexpression that activates quiescent cells with the ability to differentiate into cardiomyocytes and vascular cells.8,16 This temporary overexpression of low potency and short duration is sufficient to cause an invasion from the surface to the area of the heart wall injured by AMI. This facilitates the restoration of damaged tissues and the formation of new vessels. However, it does not fully compensate for the damage caused.18
As mentioned at the start of this chapter, the transcription factor WT1 was originally described in renal cancer (nephroblastoma). Its primary function is the activation of multiple genes and it is also involved in the post-transcriptional processes of numerous organs. With particular relevance for the heart, WT1 expression begins on the epicardial side and expands towards the luminal site of the heart, modulating the expression of vessel precursor cells.19 However, and as mentioned above, WT1 is re-expressed throughout the pericardium within 24h of the onset of AMI. WT1 is initially observed in the endothelial cells of the region bordering the infarcted area, and later in the infarcted segment with peak expression on day 7 post-AMI, before returning to baseline levels after three months. In this regard and in answer to the question what could cause this change in expression, the inflammatory route emerges as a potential trigger of WT1 expression activation post-AMI. This hypothesis is supported by studies that demonstrate that pro-inflammatory cytokines like TNF-α, IL-1β and IL-6 positively regulate WT16 (Fig. 1).

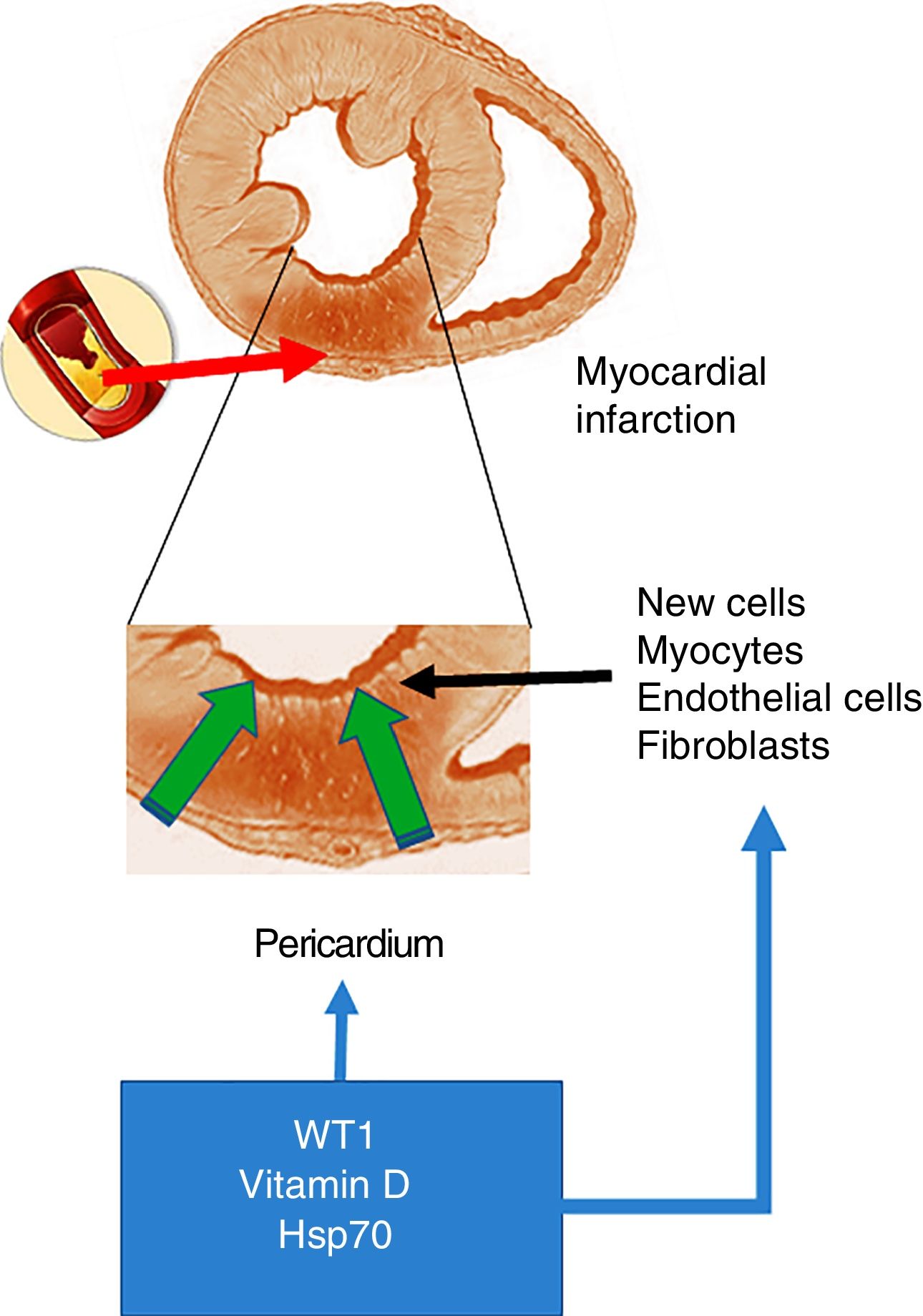
Acute myocardial infarction. The top of the figure shows an occlusion of the coronary artery, which triggers the development of an area of acute myocardial infarction. The enlarged image in the middle shows that this event gives rise to a necrotic area, which promotes the activation of cells with the ability to regenerate. The bottom of the figure shows the cells that invade the infarcted ventricular wall from the pericardium. These cells, together with those with the ability to regenerate, could be positively modulated by WT1, vitamin D levels and Hsp70.
Furthermore, WT1 expression in the epicardium after injury may also be induced by soluble factors released by the myocardium into the pericardial fluid, where some of the responses are mediated by vascular endothelial growth factor (VEGF), the protein thymosin beta-4 (thymosin β4) and prokineticins. This signalling pathway may be activated by various mediators, including vitamin D, which in turn may stimulate WT1 expression. In this regard, it has been reported that vitamin D receptors (VDR) are transcriptionally modulated by WT1.19
Vitamin D is naturally fat-soluble and is now considered to be an actual hormone. The body gets vitamin D through the diet and/or exposure to sunlight. Its metabolism may vary and is particularly susceptible to diet, geographical region, latitude, season, cultural habits and stress. Its main actions are mediated by binding to the VDR, thereby regulating the transcription of more than 200 genes. The VDR can be found in almost all the organs, including the heart.10 In light of the above, it is interesting to note that 50% of all heart disease involves vitamin D deficiency. Hypovitaminosis D is highly prevalent in the general population at 30–50% and is associated with increased cardiovascular mortality, AMI, arterial hypertension and diabetes, with a common denominator of inflammation.13 The use of paricalcitol (a vitamin D analogue) has been shown to reduce cardiovascular mortality. The positive cardiovascular effects are seen in the reduced expression of genes that cause calcification, reduced oxidative stress and reduced calcium and phosphorus absorption by the intestines. Consistent with the above, patients with low vitamin D levels are recommended to take supplements, taking into account parathyroid hormone (PTH) concentrations to avoid secondary hyperparathyroidism.
With particular focus on the inflammatory signalling pathway, it has been reported that paricalcitol reduces levels of IL-1, IL-6 and TNF-α. Furthermore, there is growing evidence from basic and clinical trials of the close relationship between cardiovascular disease (CVD), vitamin D deficiency and the stimulation of the renin–angiotensin system (RAS). More specifically, in addition to its traditional function on the metabolism of phosphorus and calcium, vitamin D also exerts multiple immunoregulatory and cytoprotective actions as a result of inhibition of the RAS system.20 Vitamin D binds to various proteins and reduces post-AMI myocardial injury, where expression of the VDR in normal myocardium is generally minimal, but which increases during left ventricular hypertrophy. As such, treatment with vitamin D seems to have protective potential to improve diabetic cardiomyopathy by modulating VDR signalling, partly through the PARP1/SIRT1/mTOR pathway.21 Specifically, SIRT1 enzymatically potentiates vitamin D signalling through deacetylation of VDR. This is of particular interest as it is known that SIRT1 is an enzyme with deacetylase activity that contributes to cell viability.22 In contrast, mTOR is regulated by levels of vitamin D and its receptors. mTOR is a protein with serine/threonine kinase activity and is an essential component of complexes conserved in eukaryotes. The protein mTOR regulates a wide variety of processes, including growth, proliferation, motility, transcription and translation.23 Vitamin D levels may also regulate the activity of PARP1,24 a protein that belongs to a family of polymerases that are involved in a large number of cellular processes, primarily DNA repair and programmed cell death.
In light of the above, the impact of vitamin D deficiency on the onset of hypertrophy and myocardial inflammation is clear. At the same time, a number of investigations have shown elevated levels of heat shock proteins (HSP) in patients with systemic hypertension, coronary artery disease, carotid atherosclerosis, myocardial infarction and ischaemia.25 This is relevant as one of these, Hsp70, with a molecular weight of 70kDa, may interact with VDR before it is activated by binding to its endogenous ligand, vitamin D.26 This mechanism suggests that the inflammatory process may raise Hsp70 levels, and this could interact with VDR to the benefit of the myocardium.
HSPs are a group of proteins that were originally thought to be activated by an increase in temperature.27 However, it is now known that they are also induced by many other factors, including the cold, stress, hypoxia and inflammation. They have been exhaustively classified in accordance with their molecular weight. With regard to Hsp70 specifically, the investigations conducted have shown that this family of proteins plays a role in the folding and assembly of newly synthesised proteins, the folding of aggregated proteins, membrane translocation, secretory proteins and in controlling the regulatory activity of multiple structural and functional proteins. Hsp70 are also produced in large quantities by cells in response to mechanical or ischaemic stress and stimulation with cytosines. They act as a highly conserved ancestral protective mechanism against various adverse conditions. Within the cell, HSPs act as true molecular chaperones, facilitating correct protein assembly as well as the translocation of oligomers, but they also promote the elimination of irreversibly damaged proteins. They are activated by pro-inflammatory cytokines and regulate apoptosis.28–30
The modulatory effect of these chaperones may help to regulate various processes associated with atherosclerosis, like oxidative stress, inflammation and apoptosis (Fig. 2A). This regulation could promote the positive regulation of WT1 as an associated factor in the signalling pathway to reduce the harmful effects of remodeling. In this sense, Hsp70 levels seem to exert a protective effect against ischaemia. Increased Hsp70 expression improves ischaemic heart recovery. Because Hsp70 acts as a key cofactor for WT1 function, this chaperone may play a key role during renal differentiation. This same mechanism has also been proposed very recently in the heart. It has been shown that WT1 expression is modulated by nitric oxide (NO) availability.31 This is relevant as it is known that NO levels condition Hsp70 expression, and it can be postulated that Hsp70 acts as a central factor in regulating WT1 expression during myocardial remodeling, promoting the expression and formation of new vessels in the infarcted area. This hypothesis is supported by the original finding that describes the importance of Hsp70 for WT1 functionality17 (Fig. 2B).
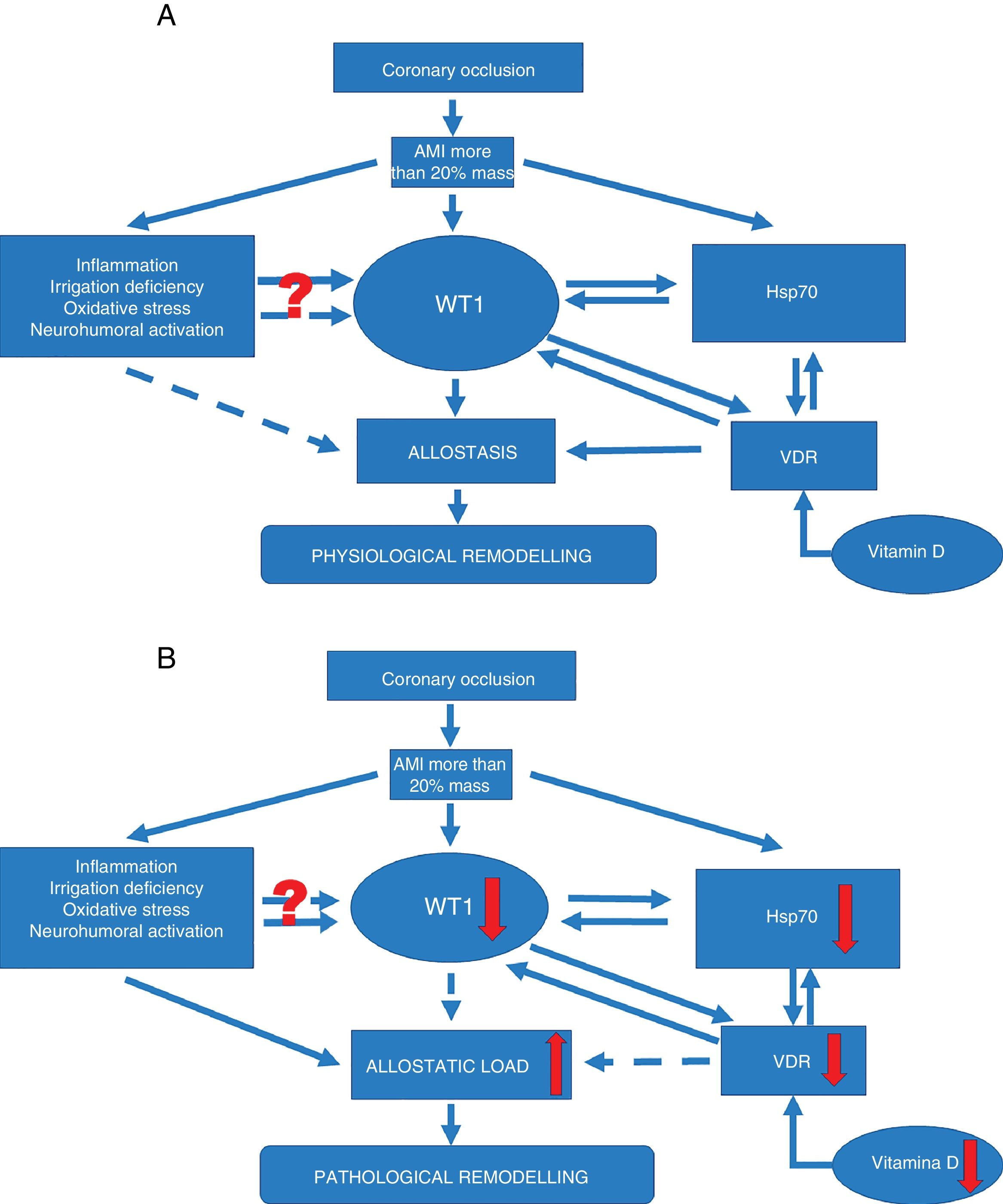
 Signalling pathway in physiological ventricular remodeling. A coronary occlusion that gives rise to an infarcted area greater than or equal to 20% could trigger allostatic-physiological remodeling. This mechanism involves known factors, including inflammation, tissue hypoxia and oxidative stress. But it also induces the expression of lesser-known factors such as transcription factor WT1 and stress response protein Hsp70. In addition, another little-known effector—vitamin D and its receptors—could promote the actions of Hsp70 and WT1 and consequently allostatic-physiological remodeling. It is still not fully understood how inflammation, irrigation deficiency, oxidative stress and neurohumoral activation might modulate WT1 expression, or why this effect is only temporary. The solid arrows represent induction, while the dotted arrows indicate inhibition. Allostasis is defined as the biological change mechanisms that exert a greater change than those mechanisms regulated by homeostasis. These mechanisms are temporarily capable of adapting to a certain instability such as that seen during AMI. (B) Signalling pathway in pathophysiological ventricular remodeling. A coronary occlusion that gives rise to an infarcted area greater than or equal to 20% could trigger pathophysiological remodeling due to allostatic load or overload. This pathological mechanism involves known factors, including inflammation, tissue hypoxia and oxidative stress. As with physiological remodeling, pathophysiological remodeling also involves lesser-known mechanisms but to a lesser extent. Reduced expression of transcription factor WT1 and the stress response protein Hsp70. Another little-known effector—vitamin D and its receptors—is also involved; alterations that promote the onset of pathophysiological remodeling. Once again it must be mentioned that it is still not fully understood how inflammation, irrigation deficiency, oxidative stress and neurohumoral activation might modulate WT1 expression, or why this effect is only temporary. The solid arrows represent induction, while the dotted arrows indicate inhibition. Allostatic load is defined as the mechanisms involved in the allostatic response, activated in such a time and manner as to facilitate effective adaptation. However, if they are not inactivated in an appropriate time and manner, they can damage the body and the systems involved in allostasis. These adaptation mechanisms up-regulated by mechanisms that have still not been fully understood during AMI instability could cause so-called pathophysiological remodeling due to allostatic overload.")
Signalling pathways. (A) Signalling pathway in physiological ventricular remodeling. A coronary occlusion that gives rise to an infarcted area greater than or equal to 20% could trigger allostatic-physiological remodeling. This mechanism involves known factors, including inflammation, tissue hypoxia and oxidative stress. But it also induces the expression of lesser-known factors such as transcription factor WT1 and stress response protein Hsp70. In addition, another little-known effector—vitamin D and its receptors—could promote the actions of Hsp70 and WT1 and consequently allostatic-physiological remodeling. It is still not fully understood how inflammation, irrigation deficiency, oxidative stress and neurohumoral activation might modulate WT1 expression, or why this effect is only temporary. The solid arrows represent induction, while the dotted arrows indicate inhibition. Allostasis is defined as the biological change mechanisms that exert a greater change than those mechanisms regulated by homeostasis. These mechanisms are temporarily capable of adapting to a certain instability such as that seen during AMI.
(B) Signalling pathway in pathophysiological ventricular remodeling. A coronary occlusion that gives rise to an infarcted area greater than or equal to 20% could trigger pathophysiological remodeling due to allostatic load or overload. This pathological mechanism involves known factors, including inflammation, tissue hypoxia and oxidative stress. As with physiological remodeling, pathophysiological remodeling also involves lesser-known mechanisms but to a lesser extent. Reduced expression of transcription factor WT1 and the stress response protein Hsp70. Another little-known effector—vitamin D and its receptors—is also involved; alterations that promote the onset of pathophysiological remodeling. Once again it must be mentioned that it is still not fully understood how inflammation, irrigation deficiency, oxidative stress and neurohumoral activation might modulate WT1 expression, or why this effect is only temporary. The solid arrows represent induction, while the dotted arrows indicate inhibition. Allostatic load is defined as the mechanisms involved in the allostatic response, activated in such a time and manner as to facilitate effective adaptation. However, if they are not inactivated in an appropriate time and manner, they can damage the body and the systems involved in allostasis.
These adaptation mechanisms up-regulated by mechanisms that have still not been fully understood during AMI instability could cause so-called pathophysiological remodeling due to allostatic overload.
Finally, a very recent study conducted on the hearts of zebrafish on microRNA involved in the activation of the epicardium during heart regeneration evaluated WT1 as an epicardial marker, Hsp70 as a chaperone activated during regeneration and cardiac troponin T (cTnT), a marker of differentiated cardiomyocytes. The results revealed that WT1 and Hsp70 mark the cardiac regeneration site two to three days after ventricular resection, which underlines its importance and potential interaction.32
Conclusions and perspectivesVentricular remodeling is a component of AMI and gives rise to changes that increase long-term morbidity and mortality. The early restoration of blood flow, as well as the use of beta-blockers, angiotensin-converting enzyme inhibitors and statins, have all been proven effective. Our knowledge of the mechanisms involved in the development of potential treatments that aim to replace lost cardiac muscle cells has improved in recent years. Specifically, WT1, its relationship with the pericardium and the formation of cells for new vessels could be a new treatment pathway that ensures cell regeneration over time and thereby minimises long-term progression, favouring this group of patients. In addition, the use of vitamin D is one of the tools that is postulated could activate cell regeneration for longer than WT1 through the VDR and its association with Hsp70, giving rise to physiological and favourable remodeling. Further study of the pathophysiological mechanisms involved during AMI will facilitate the development of new strategies for the treatment of complications and a more aggressive approach, with the intention of preventing, attenuating or even reversing the progressive damage caused during post-AMI pathophysiological remodeling.
FundingWe would like to thank the following for their support in the research, drafting and publishing of this article: The Research and Technology Council of the Universidad de Cuyo (SECyT), Mendoza, Argentina and the Agencia Nacional de Promoción Científica y Tecnológica [National Agency for Scientific and Technological Promotion] (ANPCyT); both awarded to Walter Manucha. Grant PICT 2016-4541.
Conflicts of interestNone of the authors declare any conflicts of interest regarding the content of this article.
Please cite this article as: Sanz RL, Mazzei L, Manucha W. Implicancias del factor de transcripción WT1 asociado al remodelado cardiaco patológico postinfarto miocárdico. Clín Investig Arterioscler. 2019;31:121–127.