




The elevation of blood pressure produces specific organic lesions, including kidney and cardiac damage. On the other hand, cardiovascular disease usually leads to the development of hypertension. Thus, hypertension could be both a cause and a consequence of cardiovascular disease. Previous studies linked the lack of nitric oxide to cardiovascular abnormalities, including hypertension, arteriosclerosis, myocardial infarction, cardiac hypertrophy, diastolic heart failure, and reduced endothelium-derived hyperpolarizing factor responses, with shorter survival. The lack of this gas also leads to renal/cardiac abnormalities.
It is widely known that nephrogenic deficiency is a risk factor for kidney disease. Besides, recent evidence suggests that alterations in WT-1, a key nephrogenic factor, could contribute to the development of hypertension. Moreover, some genes involved in the development of hypertension depend on WT-1.
This knowledge makes it essential to investigate and understand the mechanisms regulating the expression of these genes during renal/cardiac development, and hypertension. As a consequence, the most in-depth knowledge of the complex aetiopathogenic mechanism responsible for the hypertensive disease will allow us to propose novel therapeutic tools.
La hipertensión produce lesiones orgánicas específicas como daño renal/cardíaco, mientras que la enfermedad cardiovascular generalmente conduce a la hipertensión. Por ello, la hipertensión sería tanto una causa como una consecuencia de la enfermedad cardiovascular. Estudios previos refieren falta de óxido nítrico con anomalías cardiovasculares como hipertensión y reducción de las respuestas del factor hiperpolarizante derivado del endotelio. La falta de este gas también conduce a anomalías renales/cardíacas.
Además, la deficiencia nefrogénicaes un factor de riesgo para la enfermedad renal.
Así, alteraciones en WT-1, un factor nefrógeno clave, podrían contribuir al desarrollo de hipertensión. Finalmente, el conocimiento más profundo del complejo mecanismo etiopatogénico responsable de la enfermedad hipertensiva nos permitirá proponer nuevas herramientas terapéuticas.
In developed countries, hypertension is considered one of the most important public health problems, affecting about one billion people worldwide causing 9.4million deaths/year (World Health Organization, 2013). Its inheritance has a multifactorial origin, just as the determination of stature and weight. Thus, multiple genes and multiple environmental factors (sodium intake, renin, insulin resistance, sleep apnea, and age, among others) determine blood pressure. These facts make hypertension complex to study. Besides, most people with high blood pressure do not have any symptoms and are not aware of their condition. Although, many changes that precede the establishment of hypertension, produce determined organic lesions. One of the principal organs affected is the kidney; were there is a close relationship between hypertension and kidney disease, which remains unclear and is a matter of significant research interest. The evidence, so far, indicates that high blood pressure is both a cause and a consequence of kidney disease.
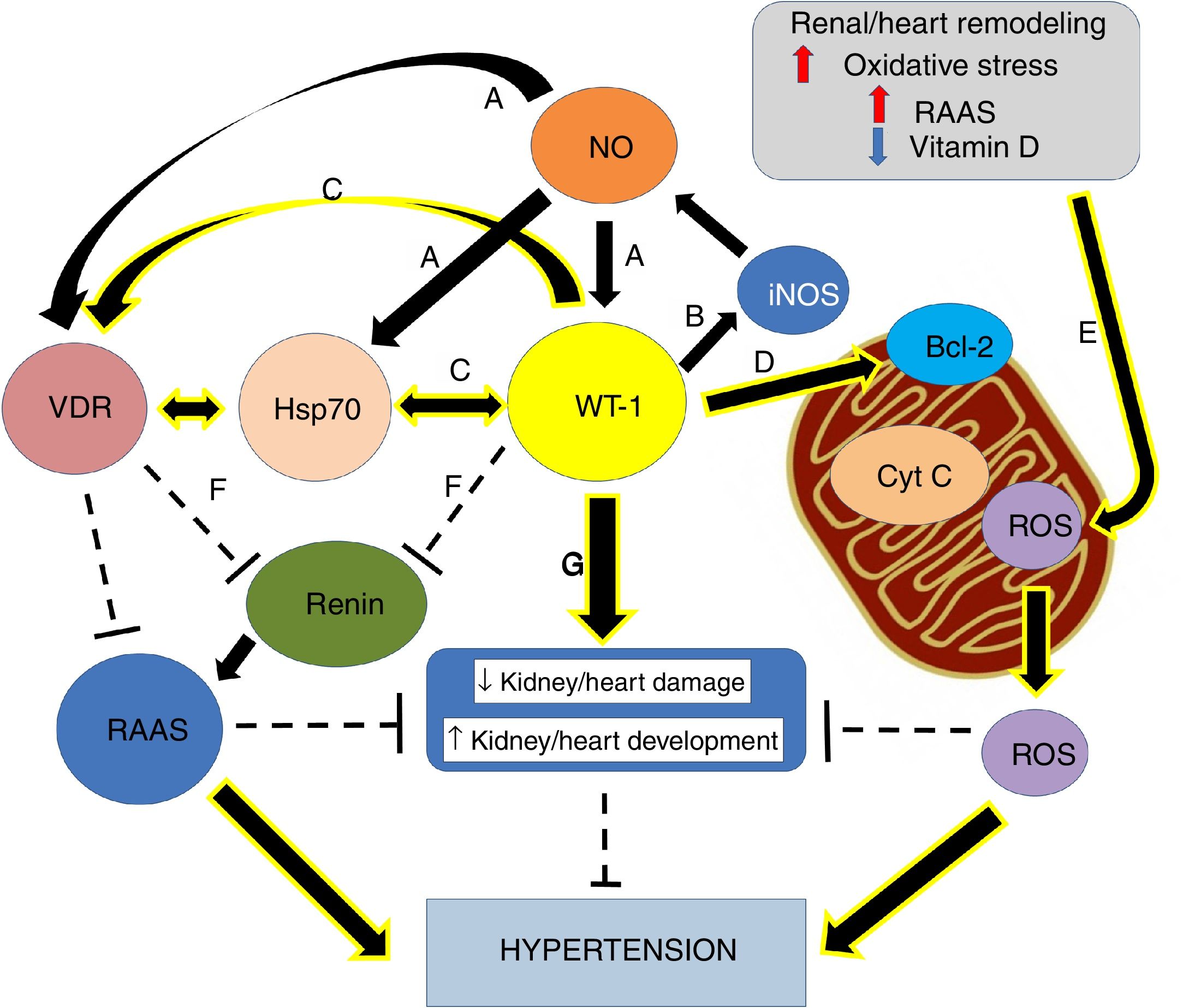
Nephrogenic disorder and low birth weight are frequently related. The first one is recognized as an essential risk factor for kidney injury, conditioning a more rapid progression of the affliction and the possible development of hypertension.1 Previous investigations concluded that maternal food limitation modifies the expression of genes related to nephrogenesis, and this fact appears to increase blood pressure. One of the disturbed genes is Wilms’ tumor transcription factor 1 (wt-1)2,3 (Fig. 1).
 Small increases in ON levels produce an increase in the expressions of Hsp70, WT-1, and VDR. (B) In turn, WT-1, as a transcription factor, regulates the expression of the principal isoform of the NO synthase, producing a positive feedback. (C) Also, WT-1 positively regulates Hsp70 and VDR. (D) WT-1 stabilizes Bcl-2 and the mitochondrial membrane, preventing the release of cytochrome C. In this way, cell death is avoided. (E) Renal remodeling, oxidative stress, the activation of the RAAS and the decrease of vitamin D, produce the release of ROS, which increases the risk of hypertension. (F) VDR and WT-1 inhibit renin, thus inhibiting the RAAS system, and hypertension. (G) WT-1 is not only crucial for kidney development but also to prevent kidney damage and high blood pressure. The continuous arrows indicate positive regulation, while the discontinuous lines indicate inhibition.")
Graphical overview. (A) Small increases in ON levels produce an increase in the expressions of Hsp70, WT-1, and VDR. (B) In turn, WT-1, as a transcription factor, regulates the expression of the principal isoform of the NO synthase, producing a positive feedback. (C) Also, WT-1 positively regulates Hsp70 and VDR. (D) WT-1 stabilizes Bcl-2 and the mitochondrial membrane, preventing the release of cytochrome C. In this way, cell death is avoided. (E) Renal remodeling, oxidative stress, the activation of the RAAS and the decrease of vitamin D, produce the release of ROS, which increases the risk of hypertension. (F) VDR and WT-1 inhibit renin, thus inhibiting the RAAS system, and hypertension. (G) WT-1 is not only crucial for kidney development but also to prevent kidney damage and high blood pressure. The continuous arrows indicate positive regulation, while the discontinuous lines indicate inhibition.
The kidneys are very complex structures; their functional units contain about 10,000 cells with at least 14 different kinds of cells. The renal morphogenesis must be entirely controlled and that the conversion of mesenchymal mesodermal cells into polarized epithelial cells4 is a critical step in this process. There is evidence that WT-1 is involved in the regulation of cell proliferation and differentiation. It binds to developmental targets mRNA, especially to 3′ untranslated regions.5 In the 90s, WT-1 expression was selectively found in the glomerular epithelium and metanephric blastemal during intrauterine development.6
The WT-1 expression patterns suggest an essential role of this gene during urogenital development. Furthermore, WT-1 was sub-located in the nucleus cell, suggesting its role as a transcription factor.7 However, Niksic et al. found that WT-1 protein is not restricted to the nucleus and shuttles continuously between the nucleus and cytoplasm.8 They revealed that, depending on the cell kind, about 10–50% of total cellular WT-1 could be identified in the cytoplasm. A substantial amount of cytoplasmic WT-1 was linked with ribonucleoprotein particles and dynamically translating polysomes, supporting its role in RNA metabolism and suggesting its participation in the regulation of translation.
Moreover, the WT-1 is necessary for the signal from the metanephric blastema that gives origin to the initial growth of the ureteric bud.7,9,10 Nevertheless, WT-1 is necessary not only for normal kidney development but also for gonads and mesothelium.11 Besides, it is necessary through normal postnatal life and reactivated during tissue repair and adult tumorigenesis.12 Martinez-Estrada et al. found that epicardial-specific knockout of WT-1 in mice led to embryonic lethality due to cardiovascular failure. In fact, Kardon et al., showed that mutations in wt-1 gene, affecting pulmonary and cardiac development.13 Thus, studies with mutant hearts revealed a derepression of the epithelial phenotype in epicardial cells and during embryonic stem cell differentiation leading to a reduced number of mesenchymal progenitor cells and their derivatives14 (Fig. 1).
WT-1 functions in the cardiovascular systemThe heart is an organ key for oxygen distribution and nutrients throughout the body; therefore, it is the first organ to develop, and it is already functional in its most primitive structure during embryogenesis. Recent studies indicate that WT-1 is essential for many aspects of cardiac development. The WT-1 expression is first observed in the proepicardium.15
The proepicardium (PE) cells are composed of two cell lineages, one of them, expressing T-box18 (Tbx18), a molecular marker of PE cells and epicardium, and WT-1 (the other expressing Scx and Semad3)16 which gave rise to smooth muscle cell, cardiomyocytes and fibroblasts.17–19 Thus, retinaldehyde dehydrogenase 2 (RALDH2), WT-1, transcription factor 21 (TCF21), and T-box18 transcription factors are highly enriched in the embryonic epicardium,17,18,20,21 and their expression is reactivated in the adult epicardium after injury.22
In this sense, Smart and Riley found in mice that stem cell population is contained in the adult heart. After myocardial infarction, this progenitor line has the potential to contribute to terminally differentiated cardiomyocytes. The skill to revive ordinarily dormant epicardium-derived cells lies in the identification of crucial stimulatory factors, such as thymosin β4 (Tβ4), and clarification of the molecular cues used in the embryo to coordinate cardiovascular development.23 They exposed an innovative genetic marker of the stimulated adult progenitors via re-expression of WT-1, through priming by Tβ4, a peptide shown to re-establish vascular potential to adult epicardium-derived progenitor cells with damage. Growing evidence showed an epicardial origin of the precursor population, and embryonic reprogramming resulted in the mobilization and differentiation of this population to give rise to de novo cardiomyocytes. They revealed that derived cardiomyocytes could structurally and functionally integrate with resident muscle. Thus, stimulation of this adult progenitor pool denoted an essential stage toward resident cell-based therapy in human ischemic heart disease.24
Also, Huang et al. set an organ culture from mouse embryonic heart and established a gene expression system that allows the identification of epicardial enhancers triggered during heart development and damage. They found that conserved regions (CRs) CR2 in Raldh2 and CR14 in WT-1 are strong epicardial enhancers that respond to CCAAT/enhancer-binding protein (C/EBP). Epicardial activation of these enhancers depends on a combinatorial transcriptional code centered on C/EBP transcription factors. Disruption of C/EBP signaling in the adult epicardium reduced injury-induced neutrophil infiltration and improved cardiac function.25
Duim et al. suggested a potential role for WT-1 in cardiac vessel formation in development and disease based on higher WT-1 expression in proliferating endothelial cells, and its regulation on Cyclin D1 expression. Thus, WT-1 knockdown inhibits the proliferation of endothelial cells, and endothelial cells lacking WT-1 are not capable of establishing a proper vascular network in vitro.26
As observed, WT-1 has numerous functions, and mutations in wt-1 gene cause a full spectrum of renal and extrarenal manifestations, it is homozygously mutated in a subset of Wilms’ tumors. Heterozygous mutations in wt-1 give rise to congenital anomalies.3 Mutations disrupting the DNA-binding domain of WT-1 produce a potentially dominant-negative phenotype. Thus, the resulting proteins physically associate with wild-type WT-1 in vivo and may result in its sequestration within subnuclear structures.27
Lipska et al. evaluated patients with the steroid-resistant nephrotic syndrome, with WT-1-related steroid-resistant nephrotic syndrome relative to WT-1-negative patients. They found a wide range of expressivity, robust genotype–phenotype associations, and high risk and significance of extrarenal complications in WT1-associated nephropathy. WT-1 patients more frequently presented with chronic kidney disease and hypertension at diagnosis and exhibited more rapid disease progression.28
In addition to the mutations in wt-1, maternal food limitation also changes the expression of key fetal gene necessary for nephrogenesis. In fact, one of the altered genes is precisely wt-1, and this seems to contribute to hypertension development.2 Thus, nephrogenic deficiency is frequently linked to low birth weight, and it has been recognized as a potent risk factor for kidney injury, leading to a faster evolution of the disease and the possible development of hypertension.1 Therefore, hypertension development is a significant consequence of the altered nephrogenic process, which additionally intensifies the risk of onset and progression of kidney disease29 (Fig. 1).
Epigenetic of WT-1WT-1 in some cases stimulates the transcription of genes that in other cases represses, depending on the cellular context. Despite WT-1 and the paired box 2 gene (Pax2) are necessary to induce mesenchymal-epithelial transition and play critical roles in the progression of nephrogenesis (Fig. 1G), they could play roles in epithelial-mesenchymal transition (EMT) in heart development, and in the remnant kidney of rats following 5/6 nephrectomy. Huang et al. revealed that WT-1 and Pax2 were re-expressed in the EMT models, and these were attended by declined expression of epithelial cells markers and enhanced expression of fibrosis markers. Thus, proposing there may be beneficial value in silencing WT-1 and Pax2 to avoid or reverse renal fibrosis in EMT.30
The transcription factor Pax2 was recognized as a critical epigenetic player in the early kidney design. Changes in the epigenomic landscapes could be beneficial for the management of chronic kidney disease stemming from diabetes or hypertension as well as Wilms’ tumor. Then, it is essential to study the epigenomic modifiers and their changes under normal physiological conditions. Growing data of epigenetics can be used for diagnostic and therapeutic purposes.31 Aiden et al., using ChIP-Seq, characterized epigenetic of Wilms’ tumor mapping the promoter chromatin states of genes in this tumor relative to the fetal and adult kidney.32 They surmised that the persistence of the bivalent chromatin code on the promoters of genes essential for kidney development disturb normal development in the Wilms’ tumor.31
Since in many WTs the wt-1 expression is reduced and only in 10–20% of WTs exist a mutation in this gene, it was appropriate to study wt-1 gene silencing due to epigenetic modifications. Dallosso et al. showed that both WT1-AS (a WT-1 antisense transcript) and AWT-1 (a novel alternative WT-1 transcript), were imprinted in the healthy kidney with expression restricted to the paternal allele. Nevertheless, Wilms’ tumor sections showed biallelic AWT-1 expression, suggesting the relaxation of AWT-1 imprinting in a subclass of WTs. They concluded that human chromosome 11p13 is an imprinted locus, which may propose a molecular basis for the strong bias of paternal allele mutations and incomplete penetrance observed in syndromes with inherited WT-1 mutations.33
Frequently, nephroblastoma histology exposes a disorganized renal development showing blastema and epithelia randomly interspersed in varying amounts of the stroma. Familial cases of Wilms’ tumor, as well as retinoblastoma, fit Knudson's “two-hit” model, according to which a germline mutation of one WT-1 allele predisposes to the tumor while an additional somatic mutation of the other allele causes malignant transformation. Thus, for the manifestation of the tumor, the functional damage of both alleles is needed.34
However, not all Wilms’ tumor cases fit this model because the majority of Wilms’ tumors do not show a mutation of WT-1. Some of the mechanisms recognized to date involve dominant negative WT-1 mutations, an interaction of the WT-1 gene product with other mutated transcription factors such as p53; also, loss of imprinting, and mutations of other tumor suppressor genes at 11p15 or other loci. Although classic Wilms’ tumor is associated with good prognosis (85% survival), its anaplastic form is often fatal. Thus, Wilms’ tumor remains the center of attention for further investigation because it offers opportunities for studying normal kidney development; as for understanding the molecular basis for clinically critical anaplastic forms, as well as for elucidating the molecular mechanisms of tumor suppressor genes35 and WT-1 is currently becoming even more critical due to its role in cardiovascular system. Previous studies indicated that epicardial-specific knockout of WT-1 in mice led to embryonic lethality due to cardiovascular failure. Mutant hearts showed reduced numbers of mesenchymal progenitor cells and their derivatives.14 Therefore, the study of its expression and regulation during the development of hypertension would be relevant.
WT-1/Hsp70/VDR linked to hypertensionCurrent information proposes that modifications in key nephrogenic factors, such as WT-1, could lead to hypertension development. In fact, some genes linked to hypertension development are regulated by the transcription factor WT-1.29,36 Thus, the WT-1 (-KTS) protein regulates the renin gene transcription (Fig. 1F). In this way, overexpression of WT-1 (-KTS) reduced the expression of renin gene cell cultures, while knockdown of WT-1 protein increased mRNA expression of cellular renin. It is consistent with observations in patients with inherited mutations in WT-1. They exposed increased plasma renin and hypertension. In that respect, our group showed opposing relationships between wt-1 (and related genes) expression and blood pressure levels.37
Furthermore, current hypertension studies exhibited exaltation of the renin–angiotensin–aldosterone system (RAAS) linked to low vitamin D levels.38–42 In close connection, our laboratory studied the losartan effect, an angiotensin II type 1 receptor (AT1) antagonist, on the expression/localization of heat shock protein 70 (Hsp70) in primary cultures of proximal tubular cells of spontaneously hypertensive rats (SHR). We observed a cytoprotective effect of Hsp70 against hypertension induced by angiotensin II.43 In fact, membrane translocation of Hsp70 could have a protective effect by reducing NADPH oxidase activity and expression. In line with these studies, experiments with adults SHRs revealed that structural and functional changes were reversed by vitamin D receptor (VDR) induction44; which modulated a decrease in the AT1 expression and an increase in Hsp70 levels providing renal protection.45
Vitamin D is a non-toxic inducer of Hsp70 in the rat kidney.46 Also, the lack of Hsp70 gene produces the reduction of intracellular VDR concentrations.47
It is important to mention that, in primary Wilms’ tumor samples and cultured cells with inducible expression of WT-1, WT-1 and Hsp70 are physically associated. The specific sub-nuclear clusters containing WT-1 recruited the Hsp70. Also, for binding to Hsp70, WT-1 requires an amino-terminal transactivation domain and the expression of that domain itself is enough to induce the Hsp70 expression. Replacing the Hsp70 binding domain is sufficient to restore WT-1 functional properties. These observations point to Hsp70 as an essential cofactor for the WT-1 function and suggest an essential role of Hsp70 during kidney development (Fig. 1C).48 Furthermore, it has been reported that WT-1 regulates human VDR.27 Numerous analyses proposed that, while WT-1 can select from three binding sites within the promoter of VDR, the VDR gene activation seems to happen through a single site.27 Besides, transcriptional activation of the VDR by WT-1 can mediate renal embryonic cells apoptosis in response to vitamin D.49 Thus, evidence showed, during hypertension development, a crucial role of the vitamin D in renal cell growth and differentiation (Fig. 1C). Moreover, and following our findings, maternal vitamin D deficiency accompanies changes in the expression of critical renal factors that can slow maturation of glomeruli, extending the period of nephrogenesis.50
Thus, the decoupling between Hsp70 and VDR associated with a possible reduced expression of WT-1 could be keys to hypertension development. This data supports our recent findings in the adult SHR model. We recognized a substantial anatomical–functional connection with the expression of AT1, VDR, and Hsp70 in mitochondria.45 Moreover, mitochondria are the primary generators of reactive oxygen species (ROS) in cells, which play an essential role in hypertension development.51,52 In this sense, our laboratory studied an intricate regulation in the inflammatory pathway, which includes the probable modulation of Hsp70 and WT-1 on mitochondrial signaling and the control of cytochrome C release from mitochondria by WT-1 stabilizing Bcl2 and, in this way, avoiding the apoptosis process (Fig. 1D).53 Thus, the renal damage induced by hypertension is characterized by activation of numerous injurious pathways, including renal remodeling, oxidative stress, reduced vitamin D levels, an exaltation of RAAS and apoptosis. All these process are capable of leading to mitochondrial integrity lost (Fig. 1E).51
Essential hypertension in humans can be modeled by SHR rats.54 The SHR strain was obtained by breeding Wistar-Kyoto (WKY) rats with high blood pressure.55 The hypertension is development about 6 weeks of age without physiological, pharmacological or surgical intervention; however, environmental factors affect the development of hypertension. By comparing the glomeruli of SHR to those of control strain (WKY), from birth to hypertension development, our laboratory showed ultrastructural mitochondrial injury, before hemodynamic and clinical manifestations of hypertension.37 We also showed reduced expressions of WT-1, VDR, and Hsp70 in kidneys from neonatal SHR, before the blood pressure increase. This fact is essential, considering that renal development in rats continues 10 days after birth. Thus, nephrogenesis in the neonatal rat is equivalent to that of the mid-trimester human fetus, while renal maturation in the 2-week-old rat is comparable to that of the human child.56
Nevertheless, in 14–19-day-old rats with obstructive nephropathy, renal development is compromised, and nephron number is reduced.57 Furthermore, in this obstructed rats, we found glomerular alterations and reduced nephrogenic markers, such as WT-1.58 Hence, kidney impairment is an essential episode in hypertension progress and, thus, modification of WT-1 expression and other nephrogenic genes seems to contribute to hypertension development. Also, nitric oxide (NO), bioavailability associated with Hsp70 interaction may modulate wt-1 mRNA expression (Fig. 1A), preventing obstruction-induced cell death during neonatal unilateral ureteral obstruction. As well as, this gas could be significant by reducing hypertension.
Nitric oxide, a critical factor between nephrogenesis and hypertensionNO can either induce or inhibit apoptosis in different circumstances and depend on its concentration. Whereas excessive NO production induces cell death,59 protection against apoptosis has been shown at lower levels60 which correspond to those capable of inducing Hsp70 (Fig. 1A).61–63
An alternative antiapoptotic mechanism for NO is the induction of heats hock protein 32 (Hsp32) and Hsp70, by modification in oxidant/antioxidant cellular balance.64
Studies in our laboratory have suggested that NO can produce resistance to obstruction-induced cell death by inhibiting the intrinsic mitochondria apoptotic pathway, through the induction of Hsp70 expression. In obstructed neonatal rats, in vivo administration of L-arginine, a NO donor, induced Hsp70 expression; this was associated with cytoprotection from apoptosis and transiently decreased nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity. Opposite effects were obtained after nitro L-arginine methyl ester (L-NAME), a NO inhibitor treatment.61,62
We have reported that the apoptotic effect created by lower NO was directly associated with decreased Hsp70 expression and induction of the apoptotic signal transduction involving the activation of caspase 3 by decreasing stabilization of Bcl-2.61,62
Johannesen et al. have shown functional interactions between the gene promoter of inducible NO synthase (iNOS) and WT-1 (Fig. 1B).65 Also, a modulatory role of NO in the proliferation of T cells expressing WT-1 has been suggested.66 In fact, decreased NO and iNOS/Hsp70 expressions were associated with WT-1 low expression in obstructed kidneys.67
In addition, NO stimulates the expression of enzymes and transcription factors involved in DNA repair and modulation of apoptosis, such as the tumor suppressor p53. In turn, p53 interacts with WT-1 and modulates its ability to regulate the transcription of its respective target genes.68 Consequently, it was suggested that increased NO availability induces p53 and WT-1 mRNA expression.67 Moreover, WT-1 can stabilize p53, adjust its transactivational properties, and inhibits its ability to induce apoptosis, but, there is no effect in p53-mediated cell cycle arrest.69 In fact, WT-1 acts as a tumor suppressor gene by induction of p21 (cyclin-dependent kinase inhibitor), leading to G1 phase arrest. Nevertheless, other authors observed elevated p53 levels during obstructive nephropathy apoptosis induction.70–73 Furthermore, NO treatment preserves vascular smooth muscle cells from mitochondrial-dependent apoptosis and drives cells to quiescence through an increase in p53.74 Of special interest, p53 protein interacts with members of the Hsp70 chaperone family that can regulate its function.75,76
NO is not only important for kidney development, but also for cardiovascular development. Previous reports showed the triple NOSs null mice exhibited cardiovascular abnormalities, including hypertension, arteriosclerosis, myocardial infarction, cardiac hypertrophy, diastolic heart failure, and reduced endothelium-derived hyperpolarizing factor responses, with shorter survival. The triple NOSs null mice also displayed metabolic abnormalities, including metabolic syndrome and high-fat-diet-induced severe dyslipidemia. Furthermore, the triple NOSs null mice showed renal abnormalities (nephrogenic diabetes insipidus and pathological renal remodeling), lung abnormalities (accelerated pulmonary fibrosis), and bone abnormalities (increased bone mineral density and bone turnover).77
At the vascular level, NO regulates vascular tone, coronary perfusion, capillary permeability and platelet aggregation and, plays an essential role in the control of angiogenesis, inflammation and vascular cell proliferation. The regulation of all these processes is complex and depends not only on NO concentrations but also on the cellular compartment where it is being generated and on the activated signaling pathway, the pathophysiological situation analyzed the cellular redox state and the presence of other cellular mediators (acetylcholine, noradrenaline, bradykinin, angiotensin, etc.).78
NO is a well-known primary regulator of vascular function, and hypertension is one of the most common chronic disorder that is considered a crucial risk factor for vascular diseases.79 In fact, L-NAME-induced hypertension is an experimental model characterized by generalized NO deficiency and progressive increase in blood pressure if prolonged. L-NAME, a structural analog of L-arginine, is metabolized by nonenzymatic hydrolysis into the active form, N omega-nitro-L-arginine, which competitively binds to endothelial NOS.80 NOS inhibition attenuates both the synthesis and metabolism of NO, the smallest gaseous intercellular signaling molecule mediating the vascular relaxation. Subsequently, NO deficiency leads to systemic vasoconstriction and hypertension.81
If oxidative stress is indeed a cause of hypertension, it sounds logical to find antioxidants to produce a decrease in blood pressure. In fact, Abdel-Rahman et al. provided evidence that a new combination therapy of Hibiscus sabdariffa and Olea europaea extracts exhibited an antihypertensive potential. This combination significantly normalized the elevated systolic and diastolic blood pressure as well as the pulse rate after 2 and 4 weeks of treatment. It was also efficient to improved kidney functions and to produce upregulation of eNOS expression.82
Conclusions and perspectivesRecent evidence suggests that some genes involved in the development of hypertension depend on WT-1, therefore as a vicious circle the alterations in WT-1 expression could contribute to the hypertensive disease. This information supports the notion that it is essential to investigate and understand the mechanisms regulating the expression of these genes during renal/heart development and hypertension.
From the information compiled here, derives the importance of a profound regulation of the mechanisms that can lead to the WT-1 modulation linked to Hsp70 and VDR expression. In parallel, there is an implication of an appropriate level of NO, which allows Hsp70, WT-1, VDR expression stimulation, to avoid kidney damage and high blood pressure.
Finally, a relationship between hypertension and cardiovascular disease remains unclear and a matter of significant interest for research; however, it is proposed as a hypothesis that essential hypertension would result as a consequence of kidney/cardiac disease with nephrogenic/cardiogenic disturbances. As a consequence, the most in-depth knowledge of the complex etiopathogenic mechanism responsible for the hypertensive disease will allow us to propose novel therapeutic tools.
Authors’ contributionsAll authors contributed to conception and design of the review, with a substantial contribution to data, analysis, and interpretation of the data, drafting of the article, and critical revision of the article for intellectual content.
Ethical responsibilitiesProtection of people and animalsThe authors declare that no experiments have been conducted on humans or animals for this research.
Confidentiality of the dataThe authors declare that patient data does not appear in this article.
Right to privacy and informed consentThe authors declare that patient data does not appear in this article.
FundingThe author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: this work was supported by grants from the Research and Technology Council of Cuyo University (SECyT), Mendoza, Argentina, and from ANPCyTFONCyT, both of which were awarded to Walter Manucha, Grant no. PICT 2016-4541.
Conflict of interestsThe authors declare no conflict of interests.