




Previous studies show that mercury exposure increases cardiovascular risk, although the underlying cellular mechanisms have still not been fully studied. The aim of this project is to study, in vascular fibroblasts (VF), the effect of HgCl2 exposure on the expression of enzymes involved in the synthesis of prostanoids and reactive oxygen species (ROS). These molecules have been shown to participate in the inflammatory response associated with cardiovascular diseases.
Material and methodsAdventitial VF cultures of Sprague-Dawley rat aortas, shown to be α-actin negative by immunofluorescence, were exposed to HgCl2 (0.05–5μg/mL) for 48h. mRNA and protein levels of cyclooxygenase-2 (COX-2), microsomal prostaglandin E synthase 1 (mPGES-1), thromboxane A2 synthase (TXAS), NADPH oxidase 1 (NOX-1), and 4 (NOX-4) where analyzed using qRT-PCR and western blot, respectively. NOX activity was determined by chemiluminescence.
ResultsHgCl2 exposure increased COX-2, mPGES-1, TXAS, and NOX-1 expression and NOX activity, and decreased NOX-4 expression. The increase in NOX-1 and COX-2 expression was abolished by the treatment with inhibitors of COX-2 (10μM celecoxib) and NOX (300μM apocynin, 0.5μM ML-171).
Conclusions1) HgCl2 increases the expression of pro-inflammatory enzymes involved in ROS and prostanoid synthesis in VF. 2) There is a reciprocal regulation between COX-2 and NOX-1 pathways. 3) These effects can contribute to explain the increase in cardiovascular risk associated to mercury.
Estudios previos muestran que la exposición a mercurio aumenta el riesgo cardiovascular, sin embargo, los mecanismos celulares subyacentes no han sido esclarecidos completamente. Nuestro objetivo es estudiar el efecto de la exposición a HgCl2 sobre la expresión de enzimas involucradas en la síntesis de prostanoides y especies reactivas de oxígeno (ROS) en fibroblastos vasculares (FV). Se ha demostrado la participación de estas moléculas en la respuesta inflamatoria asociada a enfermedades cardiovasculares.
MétodosFV de la adventicia de aorta de ratas Sprague-Dawley, caracterizados por inmunofluorescencia como negativos para α-actina, fueron estimulados con HgCl2 (0,05-5μg/ml) durante 48 horas. Se analizaron los niveles de ciclooxigenasa-2 (COX-2), prostaglandina E sintasa 1 microsomal (mPGES-1), tromboxano A2 sintasa (TXAS), NADPH oxidasa 1 (NOX-1) y 4 (NOX-4) mediante qRT-PCR y western blot, respectivamente. La actividad de NOX se determinó mediante quimioluminiscencia.
ResultadosLa exposición a HgCl2 aumentó la expresión de COX-2, mPGES-1, TXAS y NOX-1, disminuyendo la expresión de NOX-4. El tratamiento con inhibidores de COX-2 (10μM celecoxib) y NOX (300μM apocynin, 0.5μM ML-171) abolió el aumento de la expresión de NOX-1 y COX-2, respectivamente.
Conclusiones1) HgCl2 aumenta la expresión de enzimas proinflamatorias implicadas en la síntesis de ROS y prostanoides en FV. 2) Hay una regulación recíproca entre las vías de COX-2 y NOX-1. 3) Estos efectos pueden contribuir a explicar el aumento del riesgo cardiovascular asociado a la exposición al mercurio.
The exposure to different heavy metals such as mercury is a risk factor in the development of different diseases. Mercury is a very toxic environmental pollutant and can be produced by natural or artificial sources. In the environment, there are three main types of mercury: elemental mercury, inorganic mercury and organic mercury, which is the most toxic.1 It has been observed that mercury exposure is not an uncommon event.2 In fact, people who consume fish regularly in their diets can reach mercury levels around the established limit.3 The main and better known toxic effect of mercury exposure is neurotoxicity.4 However, this metal also induces cardiovascular damage by increasing the risk of hypertension, myocardial infarction, coronary heart disease, generalized atherosclerosis and renal dysfunction.5–8
Cyclooxygenases are enzymes that transform arachidonic acid into prostaglandin H2 (PGH2). There are two cyclooxygenase (COX) isoforms, while COX-1 is the constitutive form, COX-2 is the inducible one and is usually overexpressed in pathological conditions.9 A wide variety of stimuli, including mercury,10,11 can induce the vascular expression of COX-2.12,13 PGH2 is transformed by different synthases into specific prostanoids that are responsible for the final response of the vessel. For example, thromboxane A2 (TXA2) and prostaglandin E2 (PGE2) are important in vascular tone14,15 and remodeling.16,17 TXA2 is produced by TXA2 synthase (TXAS), while PGE2 is synthesized by PGE2 synthases (PGES). There are three PGES isoforms being microsomal PGES-1 (mPGES-1) inducible and the main source of PGE2 in pathological conditions.18 In fact, proinflammatory stimuli that induce COX-2 are also able to induce mPGES-1.19,20
NADPH oxidase (NOX) is a family of enzymatic complexes that produce mainly superoxide anion (O2−) as reactive oxygen species (ROS). There are other sources of O2− such as cyclooxygenases or xanthine oxidase, but only NOX synthesizes it as its main product using O2 and NADPH. Seven isoforms of NADPH oxidase have been described in mammals, composed by a catalytic core formed by NOX 1–5 and/or dual oxidase 1–2 (DUOX 1–2) and several regulatory subunits. NOX-1 and NOX-4 are expressed in the adventitia layer of arteries, but their cellular location is different21 NOX-1 is in the plasma membrane22 generating O2− in pathological conditions.23 On the other hand, NOX-4 is found in focal adhesions and in endoplasmic reticulum generating O2− and H2O2 in basal conditions.21
Oxidative stress and prostanoids derived from the inducible cyclooxygenase isoform, COX-2 and mPGES-1, are responsible for hypercontractility, endothelial dysfunction and/or vascular remodeling in cardiovascular pathologies, such as in hypertension20,24,25 by decreasing NO availability and affecting cell migration and extracellular matrix deposition, among other effects.
Previous studies have demonstrated that chronic administration of low doses of mercury to rats induced endothelial dysfunction as a result of the decreased nitric oxide bioavailability induced by increases in oxidative stress.10,26,27 In addition, the effects of mercury on ROS and COX-2-derived prostanoids have been studied in vascular smooth muscle cells (VSMC) and endothelial cells.11,26 Vascular adventitia, composed of fibroblasts, collagen and elastin fibers, is a critical regulator of vessel wall function in health and disease. Fibroblasts produce substantial amounts of ROS that appear to be involved in fibroblast proliferation, connective tissue deposition and changes in vascular tone28,29 that occur in cardiovascular diseases.
This research analyzes the effect of mercury exposure on the expression of enzymes involved in the synthesis of prostanoids and ROS in cultures of adventitial fibroblasts. Our observations increase the knowledge about the mechanisms involved in the cardiovascular toxicity of mercury.
Material and methodsCell culture and experimental protocolPrimary vascular fibroblasts (VF) were obtained from the aortas of Sprague Dawley (3 month-old) male rats. Rats were euthanized by decapitation and thoracic aortas were aseptically isolated and placed in cold F-12 (HAM) medium containing 0.1% BSA, 200U/mL of penicillin and 200μg/mL of streptomycin. Fat tissue and blood cells were removed and aortas were digested with 2mg/mL of collagenase type II (Worthington, Lakewood, USA) in the same medium during 30min at 37°C in an atmosphere of CO2 (5%). Once the adventitia was isolated, it was placed on gelatinized cell culture dishes in DMEM-F12 medium supplemented with 10% fetal calf serum containing 100U/mL of penicillin and 100μg/mL of streptomycin. After 7–10 days, cells were confluent and split with PBS/trypsin-EDTA, rinsed and seeded at a density of 30% in DMEM-F12 medium. The cell cultures were used in passage 2. All these products were purchased to Sigma Chemical Co. (St. Louis, MO, USA).
VF were seeded in 6 or 12-well plates and starved 24h before the experiments. NADPH oxidase activity, mRNA levels of NOX-1, NOX-4, COX-2, mPGES-1 and TXAS and protein expression of COX-2 were analyzed in cells incubated during 48h with different concentrations of mercury chloride (HgCl2) (Sigma Chemical Co.), specified in the Results section, replacing the medium each 24h. This incubation time was chosen in order to show the effects of a long-term (chronic) exposure to mercury. Celecoxib, apocynin and ML-171 were added 30min before HgCl2 treatment at the concentrations indicated in the Results section. None of these compounds affected basal expression of either COX-2 or NOX-1.
NADPH oxidase activityThe superoxide (O2−) production generated by NADPH oxidase activity was determined by chemiluminescence assay by the addition of a mixture of lucigenin (5μM) and NADPH (100μM) to the protein sample in a final volume of 250μL. Both products were purchased in Sigma Chemical Co. Chemiluminescence was determined in a Berthold multiwell plate luminometer (Berthold Detection Systems, Sirius, Pforzheim, Germany). The value of the area under the curve for each reading was used to quantify chemiluminescence. Values of NADPH oxidase activity were normalized with the whole protein and were expressed as fold increase over the control.
Western blot analysisAfter appropriate treatments, cells were washed once in ice-cold PBS buffer (GIBCO, California, USA), scraped and harvested in RIPA buffer containing: 50mM Tris pH 7.5, 150mM NaCl, 1mM MgCl2, 1mM EDTA, 1% Nonidet-P40 (NP-40), 0.5% deoxicolate Na, 1% sodium dodecyl sulfate (SDS), a protease inhibitor cocktail (Roche Applied Science, Barcelona, Spain) and a mix of phosphatase inhibitors (1mM orthovanadate, 20mM β-glycerophosphate, 10mM NaF from Sigma–Aldrich). Cells were centrifuged 10min at 13000rpm and supernatants were transferred into a new tube. Protein content was determined with the Lowry method (Bio-Rad) and bovine serum albumin (BSA, Sigma Chemical Co.) as standard. 30μg of protein were loaded in a 10% SDS-polyacrylamide gel and then proteins were transferred to polyvinylidene difluoride membrane (Amersham, GE Healthcare, Buckinghamshire, UK) in Tris–Glycine transfer buffer with 20% methanol in a Bio-Rad Trans-Blot Cell (Bio-Rad). Membranes were blocked with 5% skim milk 30min at room temperature before being incubated with antibodies for COX-2 (1:200; Cayman Chemical, Ann Arbor, MI, USA) and β-actin (1:50000; Sigma–Aldrich). Membranes were washed and incubated with horseradish peroxidase-coupled anti-rabbit (1:2000; Bio-Rad) for 1h at room temperature. Bands were detected using the Luminata Forte detection system (Millipore Corporation, Billerica, MA, USA). Signals on the immunoblot were quantified using a computer program (NIH ImageJ software). β-Actin expression was used as loading control.
qRT-PCR assayCOX-2, mPGES-1, TXAS, NOX-1, NOX-4 and β2-microglobulin mRNA levels were determined in VF. Total RNA was obtained using TRI Reagent (Sigma Chemical Co) according to the manufacturer's recommendations and was reverse transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) with random hexamers. qPCR for TXAS was performed using Taqman Gene Expression Assays (Rn00562160_m1, Applied Biosystems) and for COX-2, mPGES-1, NOX-1, NOX-4 and β2-microglobulin was performed using the fluorescent dye SyBRGreen (iTaq FAST SyBRGreen Supermix with ROX, Bio-Rad, USA). All qPCRs were performed in duplicate. Primers sequences are: COX-2 (FW: AAGGAGTCTGGAACATTGTGAAC; RV: CAAATGTGATCTGGACGTCAACA), mPGES-1 (FW: AGGAGTGACCCAGATGTG; RV: ATGTATCCAGGCGATGAGA), NOX-1 (FW: CGGCAGAAGGTCGTGATTA; RV: TGGAGCAGAGGTCAGAGT), NOX-4 (FW: GCCTCCATCAAGCCAAGA; RV: CCAGTCATCCAGTAGAGTGTT), and β2-microglobulin (FW: ACCCTGGTCTTTCTGGTGCTT; RV: TAGCAGTTCAGTATGTTCGGCTT). Quantification was performed on a 7500 Fast (Applied Biosystems). PCR cycles proceeded as follows: initial denaturation for 30s at 95°C, followed by 40 cycles at 95°C for 5s, 60°C for 30s. At the end of the PCR, a melting curve analysis was performed to show the specificity of the product detected. To calculate the relative index of gene expression, we employed the 2−ΔΔCt method using untreated samples as calibrator.30 β2-Microglobulin was used as housekeeping gene.
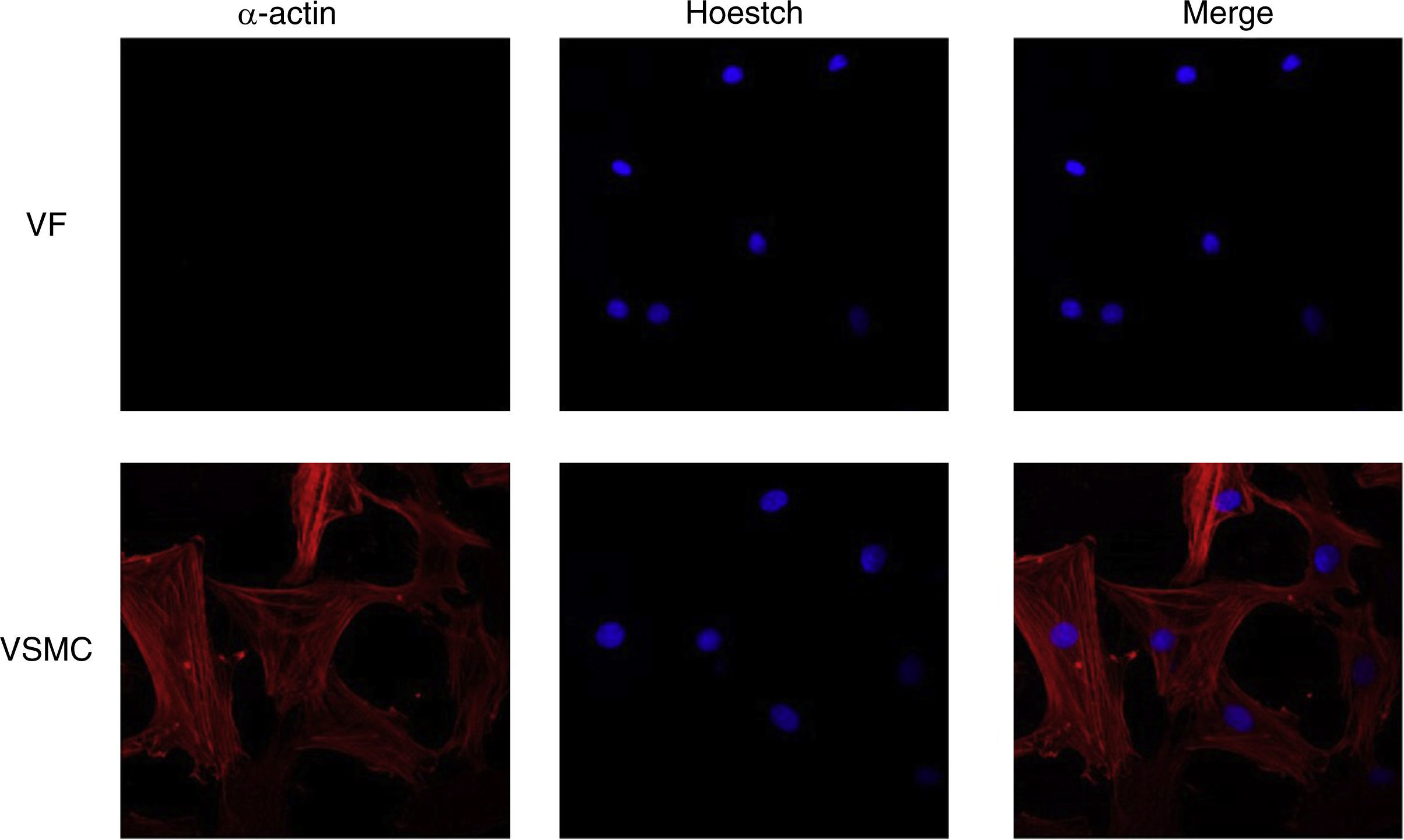
ImmunofluorescenceCells seeded in coverslips were rinsed with PBS, fixed with 4% PFA, permeabilized with 0.2% Triton-PBS, blocked with 5% BSA and subsequently incubated with mouse anti-α-actin (1/200). All these products were purchased in Sigma Chemical Co. Detection was performed with secondary antibody conjugated to Cy™3-conjugated secondary IgG (1/200; Jackson ImmunoResearch, West Grove, PA). For standard immunofluorescence microscopy, nuclei were identified by Hoechst dye 33342 (Invitrogen). Slides were examined using a Leica TS2 63X oil confocal system. Cy™3 labeled antibody was visualized by excitation at 561nm and detection at 600–700nm and Hoechst Ex 351–364nm and Em 400–500nm. The specificity of the immunostaining was evaluated by omission of the primary antibody and processed as above. Negative expression of α-actin was used to characterize VF using VSMC as positive control of staining (Fig. 1).
Drugs visualized by immunofluorescence of vascular fibroblasts (VF) and vascular smooth muscle cells (VSMC). Hoechst dye 33342 was used to stain the nuclei (blue).")
Drugs used were ML-171 and apocynin (Sigma Chemical Co). Celecoxib was generously provided by Pfizer Inc. (Groton, CT, USA). Celecoxib was dissolved in dimethylsulfoxide (DMSO), ML-171 in 90% ethanol and apocynin in distilled water.
Data analysis and statisticsStatistical analysis was performed using the Student's t test. Values were considered significant when p<0.05.
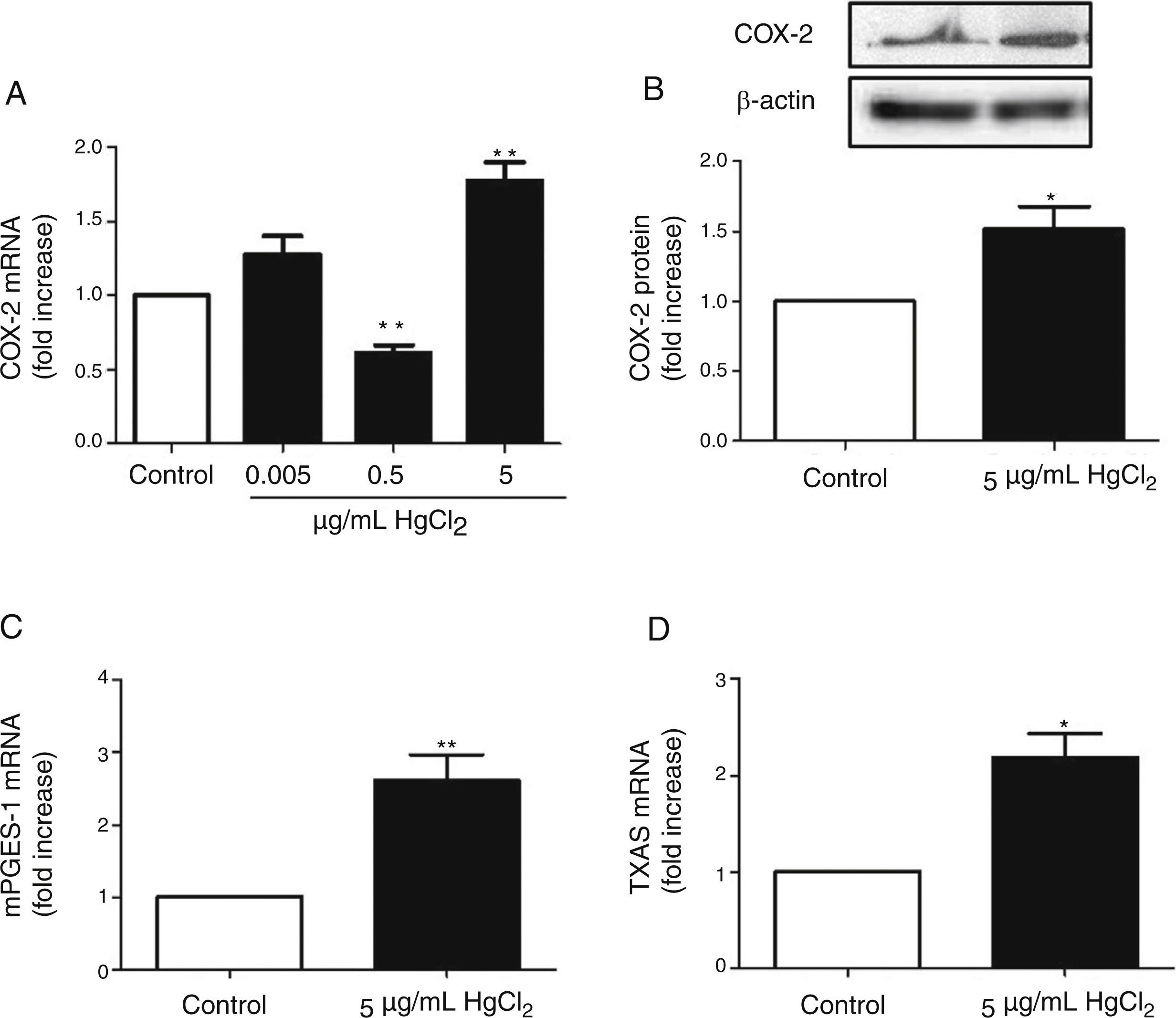
ResultsHgCl2 exposure increases COX-2, TXAS and mPGES-1 expressionIn basal conditions, rat VF showed low COX-2 mRNA and protein levels. The exposure of cells to 0.05μg/mL of HgCl2 did not affect COX-2 mRNA. However, 0.5μg/mL of HgCl2 decreased COX-2 mRNA and 5μg/mL of HgCl2 increased COX-2 mRNA and protein levels (Fig. 2A and B). In addition, HgCl2 (5μg/mL) increased TXAS and mPGES-1 mRNA expression (Fig. 2C and D).
HgCl2 increases NADPH oxidase activity and NOX-1 expression, mPGES-1 mRNA (C) and TXAS mRNA (D) levels. Data are expressed as mean±SEM. *p<0.05, **p<0.01 vs Control. n: 3–6. C: control.")
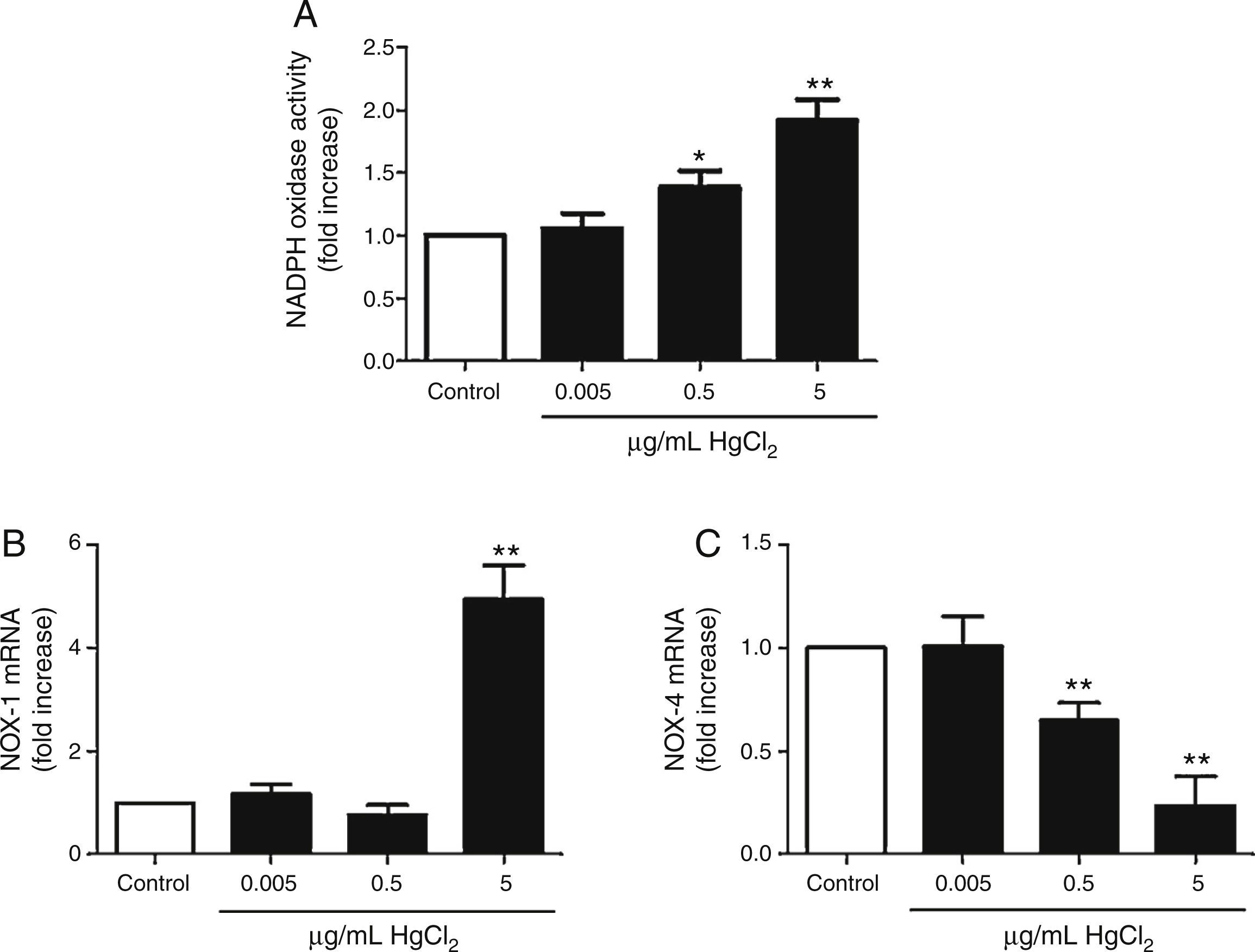
Incubation with 0.5 and 5μg/mL of HgCl2 increased concentration-dependently NOX activity, while exposure to 0.05μg/mL did not have any effect on the activity (Fig. 3A). To study if NOX activity increase was accompanied by a modulation in the NOX isoforms expression, we measured NOX-1 and NOX-4 mRNA expression. In basal conditions, rat VF showed low and high mRNA levels of NOX-1 and NOX-4, respectively. Incubation with 5μg/mL of HgCl2 increased NOX-1 mRNA, but no effect was observed with lower doses of HgCl2 (Fig. 3B). However, NOX-4 mRNA decreased in a concentration-dephendent manner with 0.5 and 5μg/mL of HgCl2 (Fig. 3C).
Reciprocal relationship between COX-2 and NOX-1 expression after HgCl2 exposure, NOX-1 (B) and NOX-4 mRNA (C) levels. Data are expressed as mean±SEM. *p<0.05, **p<0.01 vs Control. n: 5–6. C: control.")
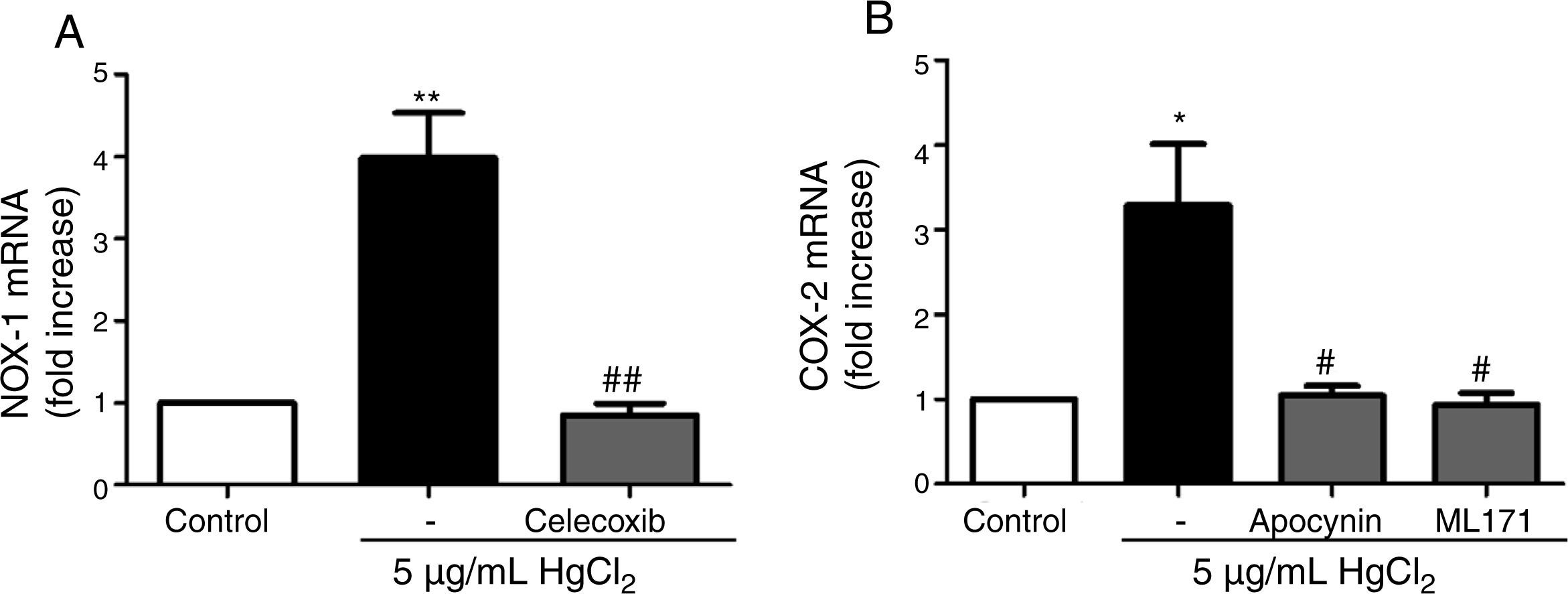
It has been previously described a relationship between COX-2 and NOX-1.11,31 Therefore, we studied this relationship in our model of mercury exposure. VF were pretreated with inhibitors of COX-2 (celecoxib, 10μM), NOX (apocynin, 300μM) and NOX-1 (ML-171, 0.5μM) and stimulated with HgCl2 (5μg/mL). The COX-2 and NOX inhibitors abolished HgCl2-induced NOX-1 and COX-2 expressions, respectively (Fig. 4).
 Effect of celecoxib (10μM) on HgCl2-induced NOX-1 expression. (B) Effect of apocynin (300μM) and ML-171 (0.5μM) on HgCl2-induced COX-2 expression. Data are expressed as mean±SEM. *p<0.05, **p<0.01 vs Control. #p<0.05, ##p<0.01 vs HgCl2. n: 6.")
NOX-1 and COX-2-derived products induce COX-2 and NOX-1 expression, respectively. (A) Effect of celecoxib (10μM) on HgCl2-induced NOX-1 expression. (B) Effect of apocynin (300μM) and ML-171 (0.5μM) on HgCl2-induced COX-2 expression. Data are expressed as mean±SEM. *p<0.05, **p<0.01 vs Control. #p<0.05, ##p<0.01 vs HgCl2. n: 6.
The results of this study demonstrate a relationship between mercury exposure and the activation of proinflammatory enzymes involved in oxidative stress and prostanoid synthesis in adventitial fibroblasts. Our experiments show that mercury induces NOX activity and COX-2, TXAS, mPGES-1 and NOX-1 expressions which supports our hypothesis that not only endothelium and VSMC but also VF are implicated in the negative cardiovascular effects and vascular remodeling caused by mercury. Previous studies have reported that these factors are closely related to cardiovascular diseases.16,17,20,21,24,25
The results of this study show that 5μg/mL of HgCl2 increase COX-2, mPGES-1 and TXAS expression. The COX-2 increase is consistent with other studies that reported a similar response to mercury exposure in VSMC11 and immune cells.32 Surprisingly, at 0.5μg/mL of HgCl2 COX-2 expression was down-regulated. The first possible explanation of this result is that COX-2 levels were only determined after 48h of stimulation, which means that mercury-dependent COX-2 levels could be increased before or after this time but not at 48h. Secondly, it is important to highlight that COX-1 isoform, although it has been described to be constitutive, can be induced by different proinflammatory stimuli.33 The COX-2 decrease observed at 0.5μg/mL could be a compensatory mechanism of a hypothetical increase in COX-1 expression. However, more experiments should be made to determine the subjacent cellular mechanisms and implications of this effect. We also observed an increase in TXAS and mPGES-1 mRNA which suggests a possible increase in TXA2 and PGE2 levels. In fact, PGE2 and TXA2 production from COX-2 was larger in aortas from chronic HgCl2-treated rats.10 It has been demonstrated that TXA2 and PGE2 participate in cell proliferation,34 migration35 and vascular remodeling.16,17 COX-2 protein was localized in adventitial and endothelial cells and that aortic COX-2 expression was greater in mercury-treated than untreated rats.10 Therefore, TXA2 and PGE2 release from adventitial fibroblasts could participate in vascular remodeling observed in rats treated with mercury.11
Mercury also induced NADPH oxidase activity in VF. The increase in NOX-1 mRNA levels observed in cells treated with mercury suggests that the increase in NOX activity is probably caused by this isoform. In agreement, it has been observed that mercury increases superoxide anion production, NADPH oxidase activity, and NOX-1 expression in VSMC.11 On the other hand, HgCl2 down-regulates NOX-4 mRNA level in VF (present results) in contrast to that observed in VSMC.11 Other groups have also reported decreases in NOX-4 levels in VSMC treated with proatherogenic stimuli such as IL-1β, thrombin and platelet-derived growth factor.36 This NOX-4 mRNA decrease could be explained as a compensatory mechanism of the increase in the NADPH oxidase activity. Although the beneficial or negative effects of NOX-4 are not yet fully understood,21 the increase in NADPH oxidase activity supports the idea that mercury has not only a negative effect on the cardiovascular system by increasing COX-2 and prostanoid synthesis but also by inducing a NOX-dependent oxidative stress pathway. Moreover, NOX-1-derived ROS have been involved in cell proliferation37 and vascular remodeling,38 suggesting a link between mercury, NOX-1 and vascular remodeling.
Another important finding of this study is the demonstration of a reciprocal relationship between COX-2 and NOX-1 pathways in VF treated with mercury, as previously described in VSMC treated with the same stimulus11 and in vessels from angiotensin II infused mice.31 This positive feedback between NOX-1 and COX-2 could exacerbate the cardiovascular damage caused by mercury. However, considering the limitations of this study, we have to mention that mercury concentrations reached in vitro cannot be directly compared to the ones reached in vivo. Because of this, although the main proinflammatory mechanisms of mercury could be similar, in vivo studies should be carried out to completely validate our model.
In summary, this study demonstrates for the first time that mercury acts as a proinflammatory stimulus in VF by inducing proteins such as COX-2 and NOX. Furthermore, the negative cardiovascular effects of mercury could be worse due to the reciprocal relationship between NOX-1 and COX-2 inductions. In this sense, our study suggests that exposure to mercury may induce vascular adventitial dysfunction and, therefore, participate in the reported cardiovascular effects of mercury.5–7 Similar results of exposure to mercury have been obtained with VSMC11 and endothelial cells26; so our study suggests that mercury induces a proinflammatory environment that could affect not only the functionality and cell properties of the intima and media layers, but also the whole vascular wall. VF are crucial in the matrix production and homeostasis of the wall of blood vessels, and ROS derived from VF are involved in important cardiovascular diseases such as atherosclerosis and hypertension among others.28 In this sense, it is our thinking that adventitia damage plays an important role in the increased risk of cardiovascular disease caused by mercury. Our findings provide new evidence that HgCl2 provokes vascular wall cells dysfunction supporting the idea that mercury should be considered an environmental risk factor for cardiovascular disease. Some studies have been focus to know if the harmful cardiovascular effects ascribed to mercury intake can be counteracted by the omega-3 fatty acids, also present in fish (one of the main dietary sources of mercury). A recent study found no evidence that mercury exposure from regular fish consumption increases cardiovascular disease risk in a population of Spanish adults with high cardiovascular disease risk and high fish consumption. This implies that the mercury content in fish does not detract from the already established cardiovascular benefits of fish consumption.39 However, other study indicate that mercury diminishes the cardiovascular protective effect of omega-3 polyunsaturated fatty acids in the diet of Inuit in Canada.40
Ethical considerationsThe experimental procedures were approved by the Animal Care and Ethical Committee of the Universidad Autónoma de Madrid according to the guidelines for ethical care of experimental animals of the European Community.
Ethical disclosuresProtection of people and animalsThe authors state that all the procedures were conducted following the ethical standards of the responsible human experimentation committee and in agreement with the World Medical Association and the Declaration of Helsinki.
Confidentiality of dataThe authors state that no patient data appears in this article.
Right to privacy and informed consentThe authors state that no patient data appears in this article.
Funding sourcesThis study was supported by grants from the Ministerio de Economía y Competitividad (SAF 2012-36400; SAF 2016-80305-P), Instituto de Salud Carlos III (Red RIC, RD12/0042/0024 and CIBER CV) and Fondo Europeo de Desarrollo Regional.
Conflict of interestsNone.
All authors have contributed to the idea and design of this study; to the collection, analysis and interpretation of the data; to the writing of the draft or to the critical review of its contents and to the approval of the final version to be published.